The Cellular and Molecular Basis of Multicellular Development in the Choanoflagellate Salpingoeca Rosetta
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Predicted Glycosyltransferases Promote Development and Prevent Spurious Cell Clumping in the Choanoflagellate S
RESEARCH ADVANCE Predicted glycosyltransferases promote development and prevent spurious cell clumping in the choanoflagellate S. rosetta Laura A Wetzel1,2, Tera C Levin1,2, Ryan E Hulett1,2, Daniel Chan1,2, Grant A King1,2, Reef Aldayafleh1,2, David S Booth1,2, Monika Abedin Sigg1,2, Nicole King1,2* 1Department of Molecular and Cell Biology, University of California, Berkeley, Berkeley, United States; 2Howard Hughes Medical Institute, University of California, Berkeley, Berkeley, United States Abstract In a previous study we established forward genetics in the choanoflagellate Salpingoeca rosetta and found that a C-type lectin gene is required for rosette development (Levin et al., 2014). Here we report on critical improvements to genetic screens in S. rosetta while also investigating the genetic basis for rosette defect mutants in which single cells fail to develop into orderly rosettes and instead aggregate promiscuously into amorphous clumps of cells. Two of the mutants, Jumble and Couscous, mapped to lesions in genes encoding two different predicted glycosyltransferases and displayed aberrant glycosylation patterns in the basal extracellular matrix (ECM). In animals, glycosyltransferases sculpt the polysaccharide-rich ECM, regulate integrin and cadherin activity, and, when disrupted, contribute to tumorigenesis. The finding that predicted glycosyltransferases promote proper rosette development and prevent cell aggregation in S. rosetta suggests a pre-metazoan role for glycosyltransferases in regulating development and preventing abnormal tumor-like multicellularity. *For correspondence: DOI: https://doi.org/10.7554/eLife.41482.001 [email protected] Competing interests: The authors declare that no competing interests exist. Introduction Funding: See page 23 The transition to multicellularity was essential for the evolution of animals from their single celled Received: 05 September 2018 ancestors (Szathma´ry and Smith, 1995). -

Lecture Notes: the Mathematics of Phylogenetics
Lecture Notes: The Mathematics of Phylogenetics Elizabeth S. Allman, John A. Rhodes IAS/Park City Mathematics Institute June-July, 2005 University of Alaska Fairbanks Spring 2009, 2012, 2016 c 2005, Elizabeth S. Allman and John A. Rhodes ii Contents 1 Sequences and Molecular Evolution 3 1.1 DNA structure . .4 1.2 Mutations . .5 1.3 Aligned Orthologous Sequences . .7 2 Combinatorics of Trees I 9 2.1 Graphs and Trees . .9 2.2 Counting Binary Trees . 14 2.3 Metric Trees . 15 2.4 Ultrametric Trees and Molecular Clocks . 17 2.5 Rooting Trees with Outgroups . 18 2.6 Newick Notation . 19 2.7 Exercises . 20 3 Parsimony 25 3.1 The Parsimony Criterion . 25 3.2 The Fitch-Hartigan Algorithm . 28 3.3 Informative Characters . 33 3.4 Complexity . 35 3.5 Weighted Parsimony . 36 3.6 Recovering Minimal Extensions . 38 3.7 Further Issues . 39 3.8 Exercises . 40 4 Combinatorics of Trees II 45 4.1 Splits and Clades . 45 4.2 Refinements and Consensus Trees . 49 4.3 Quartets . 52 4.4 Supertrees . 53 4.5 Final Comments . 54 4.6 Exercises . 55 iii iv CONTENTS 5 Distance Methods 57 5.1 Dissimilarity Measures . 57 5.2 An Algorithmic Construction: UPGMA . 60 5.3 Unequal Branch Lengths . 62 5.4 The Four-point Condition . 66 5.5 The Neighbor Joining Algorithm . 70 5.6 Additional Comments . 72 5.7 Exercises . 73 6 Probabilistic Models of DNA Mutation 81 6.1 A first example . 81 6.2 Markov Models on Trees . 87 6.3 Jukes-Cantor and Kimura Models . -

The Architecture of Cell Differentiation in Choanoflagellates And
bioRxiv preprint doi: https://doi.org/10.1101/452185; this version posted October 29, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 The architecture of cell differentiation in choanoflagellates 2 and sponge choanocytes 3 Davis Laundon1,2,6, Ben Larson3, Kent McDonald3, Nicole King3,4 and Pawel 4 Burkhardt1,5* 5 1 Marine Biological Association of the United Kingdom, The Laboratory, Citadel Hill, 6 Plymouth, PL1 2PB, United Kingdom 7 2 Plymouth University, Drake Circus, Plymouth, PL4 8AA, United Kingdom 8 3 Department of Molecular and Cell Biology, University of California, Berkeley, USA 9 4 Howard Hughes Medical Institute 10 5 Sars International Centre for Molecular Marine Biology, University of Bergen, 11 Thormohlensgate 55, 5020 Bergen, Norway 12 6 Current Affiliation: University of East Anglia, Norwich, NR4 7TJ, United Kingdom 13 14 *Correspondence [email protected] 15 16 17 18 19 20 21 22 23 24 25 bioRxiv preprint doi: https://doi.org/10.1101/452185; this version posted October 29, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 26 SUMMARY 27 Collar cells are ancient animal cell types which are conserved across the animal 28 kingdom [1] and their closest relatives, the choanoflagellates [2]. -

1 Lessons from Simple Marine Models on the Bacterial Regulation
bioRxiv preprint doi: https://doi.org/10.1101/211797; this version posted October 31, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Lessons from simple marine models on the bacterial regulation of eukaryotic development Arielle Woznicaa and Nicole Kinga,1 a Howard Hughes Medical Institute and Department of Molecular and Cell Biology, University of California, Berkeley, California 94720, United States 1 Corresponding author, [email protected]. 1 bioRxiv preprint doi: https://doi.org/10.1101/211797; this version posted October 31, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Highlights 2 - Cues from environmental bacteria influence the development of many marine 3 eukaryotes 4 5 - The molecular cues produced by environmental bacteria are structurally diverse 6 7 - Eukaryotes can respond to many different environmental bacteria 8 9 - Some environmental bacteria act as “information hubs” for diverse eukaryotes 10 11 - Experimentally tractable systems, like the choanoflagellate S. rosetta, promise to 12 reveal molecular mechanisms underlying these interactions 13 14 Abstract 15 Molecular cues from environmental bacteria influence important developmental 16 decisions in diverse marine eukaryotes. Yet, relatively little is understood about the 17 mechanisms underlying these interactions, in part because marine ecosystems are 18 dynamic and complex. -

In Defence of the Three-Domains of Life Paradigm P.T.S
van der Gulik et al. BMC Evolutionary Biology (2017) 17:218 DOI 10.1186/s12862-017-1059-z REVIEW Open Access In defence of the three-domains of life paradigm P.T.S. van der Gulik1*, W.D. Hoff2 and D. Speijer3* Abstract Background: Recently, important discoveries regarding the archaeon that functioned as the “host” in the merger with a bacterium that led to the eukaryotes, its “complex” nature, and its phylogenetic relationship to eukaryotes, have been reported. Based on these new insights proposals have been put forward to get rid of the three-domain Model of life, and replace it with a two-domain model. Results: We present arguments (both regarding timing, complexity, and chemical nature of specific evolutionary processes, as well as regarding genetic structure) to resist such proposals. The three-domain Model represents an accurate description of the differences at the most fundamental level of living organisms, as the eukaryotic lineage that arose from this unique merging event is distinct from both Archaea and Bacteria in a myriad of crucial ways. Conclusions: We maintain that “a natural system of organisms”, as proposed when the three-domain Model of life was introduced, should not be revised when considering the recent discoveries, however exciting they may be. Keywords: Eucarya, LECA, Phylogenetics, Eukaryogenesis, Three-domain model Background (English: domain) as a higher taxonomic level than The discovery that methanogenic microbes differ funda- regnum: introducing the three domains of cellular life, Ar- mentally from Bacteria such as Escherichia coli or Bacillus chaea, Bacteria and Eucarya. This paradigm was proposed subtilis constitutes one of the most important biological by Woese, Kandler and Wheelis in PNAS in 1990 [2]. -

The Closest Unicellular Relatives of Animals
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Current Biology, Vol. 12, 1773–1778, October 15, 2002, 2002 Elsevier Science Ltd. All rights reserved. PII S0960-9822(02)01187-9 The Closest Unicellular Relatives of Animals B.F. Lang,1,2 C. O’Kelly,1,3 T. Nerad,4 M.W. Gray,1,5 Results and Discussion and G. Burger1,2,6 1The Canadian Institute for Advanced Research The evolution of the Metazoa from single-celled protists Program in Evolutionary Biology is an issue that has intrigued biologists for more than a 2 De´ partement de Biochimie century. Early morphological and more recent ultra- Universite´ de Montre´ al structural and molecular studies have converged in sup- Succursale Centre-Ville porting the now widely accepted view that animals are Montre´ al, Que´ bec H3C 3J7 related to Fungi, choanoflagellates, and ichthyosporean Canada protists. However, controversy persists as to the spe- 3 Bigelow Laboratory for Ocean Sciences cific evolutionary relationships among these major P.O. Box 475 groups. This uncertainty is reflected in the plethora of 180 McKown Point Road published molecular phylogenies that propose virtually West Boothbay Harbor, Maine 04575 all of the possible alternative tree topologies involving 4 American Type Culture Collection Choanoflagellata, Fungi, Ichthyosporea, and Metazoa. 10801 University Boulevard For example, a monophyletic MetazoaϩChoanoflagel- Manassas, Virginia 20110 lata group has been suggested on the basis of small 5 Department of Biochemistry subunit (SSU) rDNA sequences [1, 6, 7]. Other studies and Molecular Biology using the same sequences have allied Choanoflagellata Dalhousie University with the Fungi [8], placed Choanoflagellata prior to the Halifax, Nova Scotia B3H 4H7 divergence of animals and Fungi [9], or even placed Canada them prior to the divergence of green algae and land plants [10]. -

Phylogenetics Topic 2: Phylogenetic and Genealogical Homology
Phylogenetics Topic 2: Phylogenetic and genealogical homology Phylogenies distinguish homology from similarity Previously, we examined how rooted phylogenies provide a framework for distinguishing similarity due to common ancestry (HOMOLOGY) from non-phylogenetic similarity (ANALOGY). Here we extend the concept of phylogenetic homology by making a further distinction between a HOMOLOGOUS CHARACTER and a HOMOLOGOUS CHARACTER STATE. This distinction is important to molecular evolution, as we often deal with data comprised of homologous characters with non-homologous character states. The figure below shows three hypothetical protein-coding nucleotide sequences (for simplicity, only three codons long) that are related to each other according to a phylogenetic tree. In the figure the nucleotide sequences are aligned to each other; in so doing we are making the implicit assumption that the characters aligned vertically are homologous characters. In the specific case of nucleotide and amino acid alignments this assumption is called POSITIONAL HOMOLOGY. Under positional homology it is implicit that a given position, say the first position in the gene sequence, was the same in the gene sequence of the common ancestor. In the figure below it is clear that some positions do not have identical character states (see red characters in figure below). In such a case the involved position is considered to be a homologous character, while the state of that character will be non-homologous where there are differences. Phylogenetic perspective on homologous characters and homologous character states ACG TAC TAA SYNAPOMORPHY: a shared derived character state in C two or more lineages. ACG TAT TAA These must be homologous in state. -

Burmese Amber Taxa
Burmese (Myanmar) amber taxa, on-line supplement v.2021.1 Andrew J. Ross 21/06/2021 Principal Curator of Palaeobiology Department of Natural Sciences National Museums Scotland Chambers St. Edinburgh EH1 1JF E-mail: [email protected] Dr Andrew Ross | National Museums Scotland (nms.ac.uk) This taxonomic list is a supplement to Ross (2021) and follows the same format. It includes taxa described or recorded from the beginning of January 2021 up to the end of May 2021, plus 3 species that were named in 2020 which were missed. Please note that only higher taxa that include new taxa or changed/corrected records are listed below. The list is until the end of May, however some papers published in June are listed in the ‘in press’ section at the end, but taxa from these are not yet included in the checklist. As per the previous on-line checklists, in the bibliography page numbers have been added (in blue) to those papers that were published on-line previously without page numbers. New additions or changes to the previously published list and supplements are marked in blue, corrections are marked in red. In Ross (2021) new species of spider from Wunderlich & Müller (2020) were listed as being authored by both authors because there was no indication next to the new name to indicate otherwise, however in the introduction it was indicated that the author of the new taxa was Wunderlich only. Where there have been subsequent taxonomic changes to any of these species the authorship has been corrected below. -

Bacterial Cues Regulate Multicellular Development and Mating in the Choanoflagellate, S
Bacterial cues regulate multicellular development and mating in the choanoflagellate, S. rosetta By Arielle Woznica A dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy in Molecular and Cell Biology in the Graduate Division of the University of California, Berkeley Committee in charge: Professor Nicole King, Chair Professor Russell Vance Professor Diana Bautista Professor Brian Staskawicz Spring 2017 Abstract Bacterial cues regulate multicellular development and mating in the choanoflagellate, S. rosetta By Arielle Woznica Doctor of Philosophy in Molecular and Cell Biology University of California, Berkeley Professor Nicole King, Chair Animals first diverged from their unicellular ancestors in oceans dominated by bacteria, and have lived in close association with bacteria ever since. Interactions with bacteria critically shape diverse aspects of animal biology today, including developmental processes that were long thought to be autonomous. Yet, the multicellularity of animals and the often-complex communities of bacteria with which they are associated make it challenging to characterize the mechanisms underlying many bacterial-animal interactions. Thus, developing experimentally tractable host-microbe model systems will be essential for revealing the molecules and mechanisms by which bacteria influence animal development. The choanoflagellate Salpingoeca rosetta, one of the closest living relatives of animals, has emerged as an attractive model for studying host-microbe interactions. Like all choanoflagellates, S. rosetta feeds on bacteria; however, we have found that interactions between S. rosetta and bacteria extend beyond those of predator and prey. In fact, two key transitions in the life history of S. rosetta, multicellular “rosette” development and sexual reproduction, are regulated by environmental bacteria. -
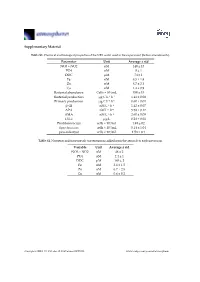
Supplementary Material Parameter Unit Average ± Std NO3 + NO2 Nm
Supplementary Material Table S1. Chemical and biological properties of the NRS water used in the experiment (before amendments). Parameter Unit Average ± std NO3 + NO2 nM 140 ± 13 PO4 nM 8 ± 1 DOC μM 74 ± 1 Fe nM 8.5 ± 1.8 Zn nM 8.7 ± 2.1 Cu nM 1.4 ± 0.9 Bacterial abundance Cells × 104/mL 350 ± 15 Bacterial production μg C L−1 h−1 1.41 ± 0.08 Primary production μg C L−1 h−1 0.60 ± 0.01 β-Gl nM L−1 h−1 1.42 ± 0.07 APA nM L−1 h−1 5.58 ± 0.17 AMA nM L−1·h−1 2.60 ± 0.09 Chl-a μg/L 0.28 ± 0.01 Prochlorococcus cells × 104/mL 1.49 ± 02 Synechococcus cells × 104/mL 5.14 ± 1.04 pico-eukaryot cells × 103/mL 1.58 × 0.1 Table S2. Nutrients and trace metals concentrations added from the aerosols to each mesocosm. Variable Unit Average ± std NO3 + NO2 nM 48 ± 2 PO4 nM 2.4 ± 1 DOC μM 165 ± 2 Fe nM 2.6 ± 1.5 Zn nM 6.7 ± 2.5 Cu nM 0.6 ± 0.2 Atmosphere 2019, 10, 358; doi:10.3390/atmos10070358 www.mdpi.com/journal/atmosphere Atmosphere 2019, 10, 358 2 of 6 Table S3. ANOVA test results between control, ‘UV-treated’ and ‘live-dust’ treatments at 20 h or 44 h, with significantly different values shown in bold. ANOVA df Sum Sq Mean Sq F Value p-value Chl-a 20 H 2, 6 0.03, 0.02 0.02, 0 4.52 0.0634 44 H 2, 6 0.02, 0 0.01, 0 23.13 0.002 Synechococcus Abundance 20 H 2, 7 8.23 × 107, 4.11 × 107 4.11 × 107, 4.51 × 107 0.91 0.4509 44 H 2, 7 5.31 × 108, 6.97 × 107 2.65 × 108, 1.16 × 107 22.84 0.0016 Prochlorococcus Abundance 20 H 2, 8 4.22 × 107, 2.11 × 107 2.11 × 107, 2.71 × 106 7.77 0.0216 44 H 2, 8 9.02 × 107, 1.47 × 107 4.51 × 107, 2.45 × 106 18.38 0.0028 Pico-eukaryote -

Predatory Flagellates – the New Recently Discovered Deep Branches of the Eukaryotic Tree and Their Evolutionary and Ecological Significance
Protistology 14 (1), 15–22 (2020) Protistology Predatory flagellates – the new recently discovered deep branches of the eukaryotic tree and their evolutionary and ecological significance Denis V. Tikhonenkov Papanin Institute for Biology of Inland Waters, Russian Academy of Sciences, Borok, 152742, Russia | Submitted March 20, 2020 | Accepted April 6, 2020 | Summary Predatory protists are poorly studied, although they are often representing important deep-branching evolutionary lineages and new eukaryotic supergroups. This short review/opinion paper is inspired by the recent discoveries of various predatory flagellates, which form sister groups of the giant eukaryotic clusters on phylogenetic trees, and illustrate an ancestral state of one or another supergroup of eukaryotes. Here we discuss their evolutionary and ecological relevance and show that the study of such protists may be essential in addressing previously puzzling evolutionary problems, such as the origin of multicellular animals, the plastid spread trajectory, origins of photosynthesis and parasitism, evolution of mitochondrial genomes. Key words: evolution of eukaryotes, heterotrophic flagellates, mitochondrial genome, origin of animals, photosynthesis, predatory protists, tree of life Predatory flagellates and diversity of eu- of the hidden diversity of protists (Moon-van der karyotes Staay et al., 2000; López-García et al., 2001; Edg- comb et al., 2002; Massana et al., 2004; Richards The well-studied multicellular animals, plants and Bass, 2005; Tarbe et al., 2011; de Vargas et al., and fungi immediately come to mind when we hear 2015). In particular, several prevailing and very abun- the term “eukaryotes”. However, these groups of dant ribogroups such as MALV, MAST, MAOP, organisms represent a minority in the real diversity MAFO (marine alveolates, stramenopiles, opistho- of evolutionary lineages of eukaryotes. -

S41467-021-25308-W.Pdf
ARTICLE https://doi.org/10.1038/s41467-021-25308-w OPEN Phylogenomics of a new fungal phylum reveals multiple waves of reductive evolution across Holomycota ✉ ✉ Luis Javier Galindo 1 , Purificación López-García 1, Guifré Torruella1, Sergey Karpov2,3 & David Moreira 1 Compared to multicellular fungi and unicellular yeasts, unicellular fungi with free-living fla- gellated stages (zoospores) remain poorly known and their phylogenetic position is often 1234567890():,; unresolved. Recently, rRNA gene phylogenetic analyses of two atypical parasitic fungi with amoeboid zoospores and long kinetosomes, the sanchytrids Amoeboradix gromovi and San- chytrium tribonematis, showed that they formed a monophyletic group without close affinity with known fungal clades. Here, we sequence single-cell genomes for both species to assess their phylogenetic position and evolution. Phylogenomic analyses using different protein datasets and a comprehensive taxon sampling result in an almost fully-resolved fungal tree, with Chytridiomycota as sister to all other fungi, and sanchytrids forming a well-supported, fast-evolving clade sister to Blastocladiomycota. Comparative genomic analyses across fungi and their allies (Holomycota) reveal an atypically reduced metabolic repertoire for sanchy- trids. We infer three main independent flagellum losses from the distribution of over 60 flagellum-specific proteins across Holomycota. Based on sanchytrids’ phylogenetic position and unique traits, we propose the designation of a novel phylum, Sanchytriomycota. In addition, our results indicate that most of the hyphal morphogenesis gene repertoire of multicellular fungi had already evolved in early holomycotan lineages. 1 Ecologie Systématique Evolution, CNRS, Université Paris-Saclay, AgroParisTech, Orsay, France. 2 Zoological Institute, Russian Academy of Sciences, St. ✉ Petersburg, Russia. 3 St.