Genome-Wide Methylation Analysis of a Large Population Sample Shows
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Capacity of Long-Term in Vitro Proliferation of Acute Myeloid
The Capacity of Long-Term in Vitro Proliferation of Acute Myeloid Leukemia Cells Supported Only by Exogenous Cytokines Is Associated with a Patient Subset with Adverse Outcome Annette K. Brenner, Elise Aasebø, Maria Hernandez-Valladares, Frode Selheim, Frode Berven, Ida-Sofie Grønningsæter, Sushma Bartaula-Brevik and Øystein Bruserud Supplementary Material S2 of S31 Table S1. Detailed information about the 68 AML patients included in the study. # of blasts Viability Proliferation Cytokine Viable cells Change in ID Gender Age Etiology FAB Cytogenetics Mutations CD34 Colonies (109/L) (%) 48 h (cpm) secretion (106) 5 weeks phenotype 1 M 42 de novo 241 M2 normal Flt3 pos 31.0 3848 low 0.24 7 yes 2 M 82 MF 12.4 M2 t(9;22) wt pos 81.6 74,686 low 1.43 969 yes 3 F 49 CML/relapse 149 M2 complex n.d. pos 26.2 3472 low 0.08 n.d. no 4 M 33 de novo 62.0 M2 normal wt pos 67.5 6206 low 0.08 6.5 no 5 M 71 relapse 91.0 M4 normal NPM1 pos 63.5 21,331 low 0.17 n.d. yes 6 M 83 de novo 109 M1 n.d. wt pos 19.1 8764 low 1.65 693 no 7 F 77 MDS 26.4 M1 normal wt pos 89.4 53,799 high 3.43 2746 no 8 M 46 de novo 26.9 M1 normal NPM1 n.d. n.d. 3472 low 1.56 n.d. no 9 M 68 MF 50.8 M4 normal D835 pos 69.4 1640 low 0.08 n.d. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

Intra-Species Differences in Population Size Shape Life History and Genome Evolution
bioRxiv preprint doi: https://doi.org/10.1101/852368; this version posted December 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Intra-Species Differences in Population Size shape Life History and Genome Evolution 2 Authors: David Willemsen1, Rongfeng Cui1, Martin Reichard2, Dario Riccardo Valenzano1,3* 3 Affiliations: 4 1Max Planck Institute for Biology of Ageing, Cologne, Germany. 5 2The Czech Academy of Sciences, Institute of Vertebrate Biology, Brno, Czech Republic. 6 3CECAD, University of Cologne, Cologne, Germany. 7 *Correspondence to: [email protected] 8 Key words: life history, evolution, genome, population genetics, killifish, Nothobranchius 9 furzeri, lifespan, sex chromosome, selection, genetic drift 10 11 1 bioRxiv preprint doi: https://doi.org/10.1101/852368; this version posted December 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 12 Abstract 13 The evolutionary forces shaping life history trait divergence within species are largely unknown. 14 Killifish (oviparous Cyprinodontiformes) evolved an annual life cycle as an exceptional 15 adaptation to life in arid savannah environments characterized by seasonal water availability. The 16 turquoise killifish (Nothobranchius furzeri) is the shortest-lived vertebrate known to science and 17 displays differences in lifespan among wild populations, representing an ideal natural experiment 18 in the evolution and diversification of life history. -

CD95 Ligand - Death Factor and Costimulatory Molecule?
Cell Death and Differentiation (2003) 10, 1215–1225 & 2003 Nature Publishing Group All rights reserved 1350-9047/03 $25.00 www.nature.com/cdd Review CD95 ligand - death factor and costimulatory molecule? O Janssen*,1, J Qian1, A Linkermann1 and D Kabelitz1 Tissue and Cellular Expression of CD95L 1 Institute for Immunology, Medical Center Schleswig-Holstein, Campus Kiel, Michaelisstrasse 5, D-24105 Kiel, Germany The CD95 ligand (CD95L, Apo-1L, FasL, CD178) is a 281- * Corresponding author: O Janssen. Tel: þ 49-431-5973377; Fax: þ 49-431- amino-acid-containing type II transmembrane protein of the 5973335; E-mail: [email protected] TNF family of death factors (Figure 1).1 Its death-inducing function is best documented in the context of activation- Received 24.4.03; revised 12.6.03; accepted 20.6.03; published online 1 August 2003 induced cell death (AICD) in T cells.2 CD95L is expressed as a Edited by T Ferguson death factor in cytotoxic T lymphocytes (CTL) to kill virally infected or transformed target cells and in natural killer (NK) cells, where it is upregulated by CD16 engagement and 3 Abstract cytokines including IL-2 and IL-12. Similarly, high levels of intracellular CD95L have been detected in monocytic cells The CD95 ligand is involved as a death factor in the with an inducible release upon activation.4 Under physiologi- regulation of activation-induced cell death, establishment cal conditions, CD95L is implicated in the control of erythroid of immune privilege and tumor cell survival. In addition, differentiation,5 angiogenesis in the eye6 and skin home- 7 CD95L may serve as a costimulatory molecule for T-cell ostasis. -

Identification of Potential Key Genes and Pathway Linked with Sporadic Creutzfeldt-Jakob Disease Based on Integrated Bioinformatics Analyses
medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Identification of potential key genes and pathway linked with sporadic Creutzfeldt-Jakob disease based on integrated bioinformatics analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Abstract Sporadic Creutzfeldt-Jakob disease (sCJD) is neurodegenerative disease also called prion disease linked with poor prognosis. The aim of the current study was to illuminate the underlying molecular mechanisms of sCJD. The mRNA microarray dataset GSE124571 was downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) were screened. -

Mitoxplorer, a Visual Data Mining Platform To
mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dynamics and mutations Annie Yim, Prasanna Koti, Adrien Bonnard, Fabio Marchiano, Milena Dürrbaum, Cecilia Garcia-Perez, José Villaveces, Salma Gamal, Giovanni Cardone, Fabiana Perocchi, et al. To cite this version: Annie Yim, Prasanna Koti, Adrien Bonnard, Fabio Marchiano, Milena Dürrbaum, et al.. mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dy- namics and mutations. Nucleic Acids Research, Oxford University Press, 2020, 10.1093/nar/gkz1128. hal-02394433 HAL Id: hal-02394433 https://hal-amu.archives-ouvertes.fr/hal-02394433 Submitted on 4 Dec 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License Nucleic Acids Research, 2019 1 doi: 10.1093/nar/gkz1128 Downloaded from https://academic.oup.com/nar/advance-article-abstract/doi/10.1093/nar/gkz1128/5651332 by Bibliothèque de l'université la Méditerranée user on 04 December 2019 mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dynamics and mutations Annie Yim1,†, Prasanna Koti1,†, Adrien Bonnard2, Fabio Marchiano3, Milena Durrbaum¨ 1, Cecilia Garcia-Perez4, Jose Villaveces1, Salma Gamal1, Giovanni Cardone1, Fabiana Perocchi4, Zuzana Storchova1,5 and Bianca H. -

A Novel PLEKHA7 Interactor at Adherens Junctions
Thesis PDZD11: a novel PLEKHA7 interactor at adherens junctions GUERRERA, Diego Abstract PLEKHA7 is a recently identified protein of the AJ that has been involved by genetic and genomic studies in the regulation of miRNA signaling and cardiac contractility, hypertension and glaucoma. However, the molecular mechanisms behind PLEKHA7 involvement in tissue physiology and pathology remain unknown. In my thesis I report novel results which uncover PLEKHA7 functions in epithelial and endothelial cells, through the identification of a novel molecular interactor of PLEKHA7, PDZD11, by yeast two-hybrid screening, mass spectrometry, co-immunoprecipitation and pulldown assays. I dissected the structural basis of their interaction, showing that the WW domain of PLEKHA7 binds to the N-terminal region of PDZD11; this interaction mediates the junctional recruitment of PDZD11, identifying PDZD11 as a novel AJ protein. I provided evidence that PDZD11 forms a complex with nectins at AJ, its PDZ domain binds to the PDZ-binding motif of nectins. PDZD11 stabilizes nectins promoting the early steps of junction assembly. Reference GUERRERA, Diego. PDZD11: a novel PLEKHA7 interactor at adherens junctions. Thèse de doctorat : Univ. Genève, 2016, no. Sc. 4962 URN : urn:nbn:ch:unige-877543 DOI : 10.13097/archive-ouverte/unige:87754 Available at: http://archive-ouverte.unige.ch/unige:87754 Disclaimer: layout of this document may differ from the published version. 1 / 1 UNIVERSITE DE GENÈVE FACULTE DES SCIENCES Section de Biologie Prof. Sandra Citi Département de Biologie Cellulaire PDZD11: a novel PLEKHA7 interactor at adherens junctions THÈSE Présentée à la Faculté des sciences de l’Université de Genève Pour obtenir le grade de Doctor ès science, mention Biologie par DIEGO GUERRERA de Benevento (Italie) Thèse N° 4962 GENÈVE Atelier d'impression Repromail 2016 1 Table of contents RÉSUMÉ .................................................................................................................. -

Uncovering Adaptive Evolution in the Human Lineage Magdalena Gayà-Vidal1,2 and M Mar Albà1,3*
Gayà-Vidal and Albà BMC Genomics 2014, 15:599 http://www.biomedcentral.com/1471-2164/15/599 RESEARCH ARTICLE Open Access Uncovering adaptive evolution in the human lineage Magdalena Gayà-Vidal1,2 and M Mar Albà1,3* Abstract Background: The recent increase in human polymorphism data, together with the availability of genome sequences from several primate species, provides an unprecedented opportunity to investigate how natural selection has shaped human evolution. Results: We compared human branch-specific substitutions with variation data in the current human population to measure the impact of adaptive evolution on human protein coding genes. The use of single nucleotide polymorphisms (SNPs) with high derived allele frequencies (DAFs) minimized the influence of segregating slightly deleterious mutations and improved the estimation of the number of adaptive sites. Using DAF ≥ 60% we showed that the proportion of adaptive substitutions is 0.2% in the complete gene set. However, the percentage rose to 40% when we focused on genes that are specifically accelerated in the human branch with respect to the chimpanzee branch, or on genes that show signatures of adaptive selection at the codon level by the maximum likelihood based branch-site test. In general, neural genes are enriched in positive selection signatures. Genes with multiple lines of evidence of positive selection include taxilin beta, which is involved in motor nerve regeneration and syntabulin, and is required for the formation of new presynaptic boutons. Conclusions: We combined several methods to detect adaptive evolution in human coding sequences at a genome-wide level. The use of variation data, in addition to sequence divergence information, uncovered previously undetected positive selection signatures in neural genes. -
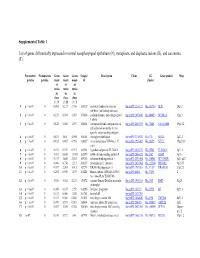
Supplemental Table 1 List of Genes Differentially Expressed In
Supplemental Table 1 List of genes differentially expressed in normal nasopharyngeal epithelium (N), metaplastic and displastic lesions (R), and carcinoma (T). Parametric Permutation Geom Geom Geom Unique Description Clone UG Gene symbol Map p-value p-value mean mean mean id cluster of of of ratios ratios ratios in in in class class class 1 : N 2 : R 3 : T 1 p < 1e-07 0 0.061 0.123 2.708 169329 secretory leukocyte protease IncytePD:2510171 Hs.251754 SLPI 20q12 inhibitor (antileukoproteinase) 2 p < 1e-07 0 0.125 0.394 1.863 163628 sodium channel, nonvoltage-gated IncytePD:1453049 Hs.446415 SCNN1A 12p13 1 alpha 3 p < 1e-07 0 0.122 0.046 1.497 160401 carcinoembryonic antigen-related IncytePD:2060355 Hs.73848 CEACAM6 19q13.2 cell adhesion molecule 6 (non- specific cross reacting antigen) 4 p < 1e-07 0 0.675 1.64 5.594 165101 monoglyceride lipase IncytePD:2174920 Hs.6721 MGLL 3q21.3 5 p < 1e-07 0 0.182 0.487 0.998 166827 nei endonuclease VIII-like 1 (E. IncytePD:1926409 Hs.28355 NEIL1 15q22.33 coli) 6 p < 1e-07 0 0.194 0.339 0.915 162931 hypothetical protein FLJ22418 IncytePD:2816379 Hs.36563 FLJ22418 1p11.1 7 p < 1e-07 0 1.313 0.645 13.593 162399 S100 calcium binding protein P IncytePD:2060823 Hs.2962 S100P 4p16 8 p < 1e-07 0 0.157 1.445 2.563 169315 selenium binding protein 1 IncytePD:2591494 Hs.334841 SELENBP1 1q21-q22 9 p < 1e-07 0 0.046 0.738 1.213 160115 prominin-like 1 (mouse) IncytePD:2070568 Hs.112360 PROML1 4p15.33 10 p < 1e-07 0 0.787 2.264 3.013 167294 HRAS-like suppressor 3 IncytePD:1969263 Hs.37189 HRASLS3 11q12.3 11 p < 1e-07 0 0.292 0.539 1.493 168221 Homo sapiens cDNA FLJ13510 IncytePD:64451 Hs.37896 2 fis, clone PLACE1005146. -

Co-Evolution of B7H6 and Nkp30, Identification of a New B7 Family Member, B7H7, and of B7's Historical Relationship with the MHC
Immunogenetics (2012) 64:571–590 DOI 10.1007/s00251-012-0616-2 ORIGINAL PAPER Evolution of the B7 family: co-evolution of B7H6 and NKp30, identification of a new B7 family member, B7H7, and of B7's historical relationship with the MHC Martin F. Flajnik & Tereza Tlapakova & Michael F. Criscitiello & Vladimir Krylov & Yuko Ohta Received: 19 January 2012 /Accepted: 20 March 2012 /Published online: 11 April 2012 # Springer-Verlag 2012 Abstract The B7 family of genes is essential in the regula- Furthermore, we identified a Xenopus-specific B7 homolog tion of the adaptive immune system. Most B7 family mem- (B7HXen) and revealed its close linkage to B2M, which we bers contain both variable (V)- and constant (C)-type have demonstrated previously to have been originally domains of the immunoglobulin superfamily (IgSF). encoded in the MHC. Thus, our study provides further proof Through in silico screening of the Xenopus genome and that the B7 precursor was included in the proto MHC. subsequent phylogenetic analysis, we found novel genes Additionally, the comparative analysis revealed a new B7 belonging to the B7 family, one of which is the recently family member, B7H7, which was previously designated in discovered B7H6. Humans and rats have a single B7H6 the literature as an unknown gene, HHLA2. gene; however, many B7H6 genes were detected in a single large cluster in the Xenopus genome. The B7H6 expression Keywords B7 family. MHC . Evolution . Natural killer patterns also varied in a species-specific manner. Human receptors . Genetic linkage . Xenopus B7H6 binds to the activating natural killer receptor, NKp30. While the NKp30 gene is single-copy and maps to the MHC in most vertebrates, many Xenopus NKp30 genes were Introduction found in a cluster on a separate chromosome that does not harbor the MHC. -

The Human Gene Connectome As a Map of Short Cuts for Morbid Allele Discovery
The human gene connectome as a map of short cuts for morbid allele discovery Yuval Itana,1, Shen-Ying Zhanga,b, Guillaume Vogta,b, Avinash Abhyankara, Melina Hermana, Patrick Nitschkec, Dror Friedd, Lluis Quintana-Murcie, Laurent Abela,b, and Jean-Laurent Casanovaa,b,f aSt. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY 10065; bLaboratory of Human Genetics of Infectious Diseases, Necker Branch, Paris Descartes University, Institut National de la Santé et de la Recherche Médicale U980, Necker Medical School, 75015 Paris, France; cPlateforme Bioinformatique, Université Paris Descartes, 75116 Paris, France; dDepartment of Computer Science, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel; eUnit of Human Evolutionary Genetics, Centre National de la Recherche Scientifique, Unité de Recherche Associée 3012, Institut Pasteur, F-75015 Paris, France; and fPediatric Immunology-Hematology Unit, Necker Hospital for Sick Children, 75015 Paris, France Edited* by Bruce Beutler, University of Texas Southwestern Medical Center, Dallas, TX, and approved February 15, 2013 (received for review October 19, 2012) High-throughput genomic data reveal thousands of gene variants to detect a single mutated gene, with the other polymorphic genes per patient, and it is often difficult to determine which of these being of less interest. This goes some way to explaining why, variants underlies disease in a given individual. However, at the despite the abundance of NGS data, the discovery of disease- population level, there may be some degree of phenotypic homo- causing alleles from such data remains somewhat limited. geneity, with alterations of specific physiological pathways under- We developed the human gene connectome (HGC) to over- come this problem. -

WO 2016/040794 Al 17 March 2016 (17.03.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/040794 Al 17 March 2016 (17.03.2016) P O P C T (51) International Patent Classification: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, C12N 1/19 (2006.01) C12Q 1/02 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, C12N 15/81 (2006.01) C07K 14/47 (2006.01) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, (21) International Application Number: KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, PCT/US20 15/049674 MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (22) International Filing Date: PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, 11 September 2015 ( 11.09.201 5) SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of regional protection available): ARIPO (BW, GH, (30) Priority Data: GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, 62/050,045 12 September 2014 (12.09.2014) US TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, (71) Applicant: WHITEHEAD INSTITUTE FOR BIOMED¬ DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, ICAL RESEARCH [US/US]; Nine Cambridge Center, LV, MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, Cambridge, Massachusetts 02142-1479 (US).