Oxidative Processes for the Direct Conversion of Coal Under Mild Conditions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Recent Advances in Direct Coal Liquefaction
Energies 2010, 3, 155-170; doi:10.3390/en3020155 OPEN ACCESS energies ISSN 1996-1073 www.mdpi.com/journal/energies Review Recent Advances in Direct Coal Liquefaction Hengfu Shui 1, Zhenyi Cai 1 and Chunbao (Charles) Xu 2,* 1 School of Chemistry and Chemical Engineering, Anhui University of Technology, Maanshan 243002, Anhui, China; E-Mails: [email protected] (H.S.); [email protected] (Z.C.) 2 Department of Chemical Engineering, Lakehead University, Thunder Bay, Ontario P7B 5E1, Canada * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-807-343-8761; Fax: +1-807-343-8928. Received: 16 November 2009 / Accepted: 19 January 2010 / Published: 27 January 2010 Abstract: The growing demand for petroleum, accompanied by the declining petroleum reserves and the concerns over energy security, has intensified the interest in direct coal liquefaction (DCL), particularly in countries such as China which is rich in coal resources, but short of petroleum. In addition to a general introduction on the mechanisms and processes of DCL, this paper overviews some recent advances in DCL technology with respect to the influencing factors for DCL reactions (temperature, solvent, pressure, atmospheres, etc.), the effects of coal pre-treatments for DCL (swelling, thermal treatment, hydrothermal treatment, etc.), as well as recent development in multi-staged DCL processes, DCL catalysts and co-liquefaction of coal with biomass. Keywords: direct coal liquefaction (DCL); processes; influencing factors; coal pre-treatment; catalysts; co-liquefaction; biomass 1. Introduction Due to the rapid increase in demand for petroleum and its declining reserves, the concern over energy security has intensified the interest in coal liquefaction, especially for those countries which are short of oil resources but have abundant coal reserves, such as the United States and China, etc. -

Liquid Coal As a Green Energy: a Review
International Journal of Engineering and Technical Research (IJETR) ISSN: 2321-0869, Volume-2, Issue-3, March 2014 LIQUID COAL AS A GREEN ENERGY: A REVIEW Saurabh Prakash, Ghanshyam Paswan & Kumar Nikhil offers significant reductions in vehicle emissions such as Abstract— Fuel market is growing more rapidly than the oxides of nitrogen, volatile organic compounds and carbon fuel production of the world. Simultaneously coal to liquid fuel mono- oxide. technology is growing and covers some of the fuel demand of the world. A cost effective technology uses the coal with low carbon value, which is not much useful in its natural form and II. METHODOLOGY the manipulated form leads to the development of liquid coal. Most of process have not been industrialized. The raw gas is The process of liquid coal production is coal to liquid or converted into liquid is an idea along with removal of sulphur, gas to liquid conversion. There are mainly two methods of phosphorous impurities which does not pollutes air so, liquid coal production. ultimately environmentally safe. A. Direct coal liquefaction process. Index terms—Bergius process, fischer- tropsch reactor, B. Indirect coal liquefaction process. liquefaction, liquefied coal, syngas. A. Direct coal liquefaction process: I. INTRODUCTION The direct coal liquefaction processes are hydrogenation Liquid coal is a manipulated component used as a fuel by and carbonisation[3]. the process of liquefaction of coal. Liquid coal is used as an alternative to oil. Coal liquefaction is particularly suited to countries that rely heavily on oil imports and have large a) Hydrogenation: domestic reserve of coal [1]. The basic method of producing Bergius process developed by Friedrich bergius in 1913 , liquid coal is gasification of coal with little oxygen and water is a method for direct conversion of coal to liquid by vapour [2]. -

Industrial Hydrocarbon Processes
Handbook of INDUSTRIAL HYDROCARBON PROCESSES JAMES G. SPEIGHT PhD, DSc AMSTERDAM • BOSTON • HEIDELBERG • LONDON NEW YORK • OXFORD • PARIS • SAN DIEGO SAN FRANCISCO • SINGAPORE • SYDNEY • TOKYO Gulf Professional Publishing is an imprint of Elsevier Gulf Professional Publishing is an imprint of Elsevier The Boulevard, Langford Lane, Kidlington, Oxford OX5 1GB, UK 30 Corporate Drive, Suite 400, Burlington, MA 01803, USA First edition 2011 Copyright Ó 2011 Elsevier Inc. All rights reserved No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means electronic, mechanical, photocopying, recording or otherwise without the prior written permission of the publisher Permissions may be sought directly from Elsevier’s Science & Technology Rights Department in Oxford, UK: phone (+44) (0) 1865 843830; fax (+44) (0) 1865 853333; email: [email protected]. Alternatively you can submit your request online by visiting the Elsevier web site at http://elsevier.com/locate/ permissions, and selecting Obtaining permission to use Elsevier material Notice No responsibility is assumed by the publisher for any injury and/or damage to persons or property as a matter of products liability, negligence or otherwise, or from any use or operation of any methods, products, instructions or ideas contained in the material herein. Because of rapid advances in the medical sciences, in particular, independent verification of diagnoses and drug dosages should be made British Library Cataloguing in Publication Data -

Coal Liquefaction
http://uu.diva-portal.org This is an author produced version of a paper published in International journal of energy research. This paper has been peer-reviewed but does not include the final publisher proof-corrections or journal pagination. Citation for the published paper: Höök, M. & Aleklett, K. ”A review on coal-to-liquid fuels and its coal consumption” International journal of energy research, 2010, Vol. 34, Issue 10: 848-864 URL: http://dx.doi.org/10.1002/er.1596 Access to the published version may require subscription. Published in International Journal of Energy Research Volume 34, Issue 10, October 2010, Pages 848-864 http://dx.doi.org/10.1002/er.1596 A review on coal to liquid fuels and its coal consumption Mikael Höök*, Kjell Aleklett* Corresponding author: [email protected], phone: +46 18-471 7643, fax: +46 18-4713513 * Uppsala University, Global Energy Systems, Department of physics and astronomy, Box 535, SE-751 21, Lägerhyddsvägen 1, Sweden, http://www.fysast.uu.se/ges Abstract Continued reliance on oil is unsustainable and this has resulted in interest in alternative fuels. Coal-to-Liquids (CTL) can supply liquid fuels and have been successfully used in several cases, particularly in South Africa. This article reviews CTL theory and technology. Understanding the fundamental aspects of coal liquefaction technologies are vital for planning and policy-making, as future CTL systems will be integrated in a much larger global energy and fuel utilization system. Conversion ratios for CTL are generally estimated to be between 1-2 barrels/ton coal. This puts a strict limitation on future CTL capacity imposed by future coal production volumes, regardless of other factors such as economics, emissions or environmental concern. -

Principles of Selectivity in Fischer-Tropsch SYNTHESIS Hans Schulz Engler-Bunte-Institute, University of Karlsruhe Kaiserstrasse 12, 76128 Karlsruhe, Germany
DGMK/SCI-Conference „Synthesis Gas Chemistry", October 4-6, 2006, Dresden, Germany Principles of Selectivity in Fischer-Tropsch SYNTHESIS Hans Schulz Engler-Bunte-Institute, University of Karlsruhe Kaiserstrasse 12, 76128 Karlsruhe, Germany Abstract The detailed selectivity of Fischer-Tropsch synthesis with iron and cobalt as catalysts with high temporal resolution has been determined and used to derive the values of probability of chain growth, chain branching and olefin/paraffin molar ratio as a function of carbon number and time. Catalyst reassembling and self-organization of the Fischer-Tropsch regime are i nvestigated. The basic principle of Fischer-Tropsch synthesis, suppression of desorption of growing chains is disclosed. This frustration governs FT-synthesis of the otherwise different systems with iron and cobalt. The advanced characterization of sites and elementary reactions (specifically with cobalt) is thought to be a more realistic basis for future theoretical calculations. Diesel fuel through Fischer-Tropsch synthesis The present interest in Fischer-Tropsch synthesis focuses on the production of Diesel fuel. Competing in coal liquefaction with hydrogenation [1], the FT-route now is superior because the fuel is very clean and exhibits excellent performance in the combustion engine (TABLE 1). However, the maximum selectivity is only ~ 40% C, as shown in Fig. 1 on the basis of calculating the theoretical product composition, assuming ideal polymerization kinetics with carbon number independent growth probability [2]. The overall Diesel selectivity can be increased to ~ 60% by running the FT-synthesis at high paraffin-wax selectivity (high growth probability pg) and mildly converting the paraffin wax through ideal hydrocracking. TABLE 1 Gas oils from direct coal liquefaction and Fischer-Tropsch synthesis Gas oil from coal hydrogenation*): Highly aromatic, Atomic H/C-ratio: 1.3 Phenols: 16 wt-% Oxygen = 3 wt.-%. -

Fischer-Tropsch Fuels from Coal, Natural Gas, and Biomass: Background and Policy
Order Code RL34133 Fischer-Tropsch Fuels from Coal, Natural Gas, and Biomass: Background and Policy Updated March 27, 2008 Anthony Andrews Specialist in Energy and Energy Infrastructure Policy Resources, Science, and Industry Division Jeffrey Logan Specialist in Energy Policy Resources, Science, and Industry Division Fischer-Tropsch Fuels from Coal, Natural Gas, and Biomass: Background and Policy Summary As the price of crude oil sets a record high, liquid transportation fuels synthesized from coal, natural gas, and biomass are proposed as one solution to reducing dependency on imported petroleum and strained refinery capacity. The technology to do so developed from processes that directly and indirectly convert coal into liquid fuel. Congress now faces decisions on whether, and to what extent, it should support such a solution. Lacking domestic petroleum resources, but abundant in coal, Germany built synthetic fuel plants during World War II that employed the Bergius coal hydrogenation process (direct liquefaction), and Fischer-Tropsch synthesis (indirect). The United States attempted to capitalize on the German experience after World War II. Despite considerable investment in synthetic fuel research and development, the United States cut support for commercialization when crude oil prices dropped and supplies stabilized in the mid-1980s. Since then, several synthetic fuels plants have been constructed around the world that convert coal, natural gas, or biomass to liquid fuels using the Fischer-Tropsch process. Several private ventures in the United States are now studying the feasibility of constructing Fischer-Tropsch synthetic fuel plants based on coal, natural gas, and biomass. Proposals to expand the use of coal to synthesize transportation fuels have generated much opposition, particularly because the carbon dioxide (CO2) produced in the Fischer-Tropsch process is a greenhouse gas associated with global warming. -

Kinetic Modelling for Coal Liquefaction Process
International Journal of Engineering Research & Technology (IJERT) ISSN: 2278-0181 Vol. 3 Issue 5, May - 2014 Kinetic Modelling for Coal Liquefaction Process Rohitbabu .G. Nalwala[1]; Pravat Kumar Rana[2]; Deepak H. Shukla[3] Chemical Engineering Department, Gharda Institute of Technology, At/Po-Lavel, Tal-Khed, Dist.-Ratnagiri, Maharashtra (India) Abstract- In 1913 Bergius developed the coal hydro the principal products can be high molecular mass fuels, liquefaction. In 1917 there were clear indication of decline in distillate fuel Oils, gasoline, or chemical feedstocks. domestic oil production and possibility of international shortage The products of both indirect and direct liquefaction have was realized. Around to be 1970 the chevron started coal higher energy content than coal. Coal typically contains 60 liquefaction process. He had designed and executed the wt% of the heating value of the liquefied products. The production of synthetic fuel, but had not given any kinetic model for monitoring the reaction. Our main focus is on to development liquids produced by the two methods are very different in a kinetic model of reaction taking place and estimating the chemical composition. Synthesis gas conversion processes kinetic parameter like rate constant, order of the reaction using give saturated hydrocarbon liquids, whereas the products of optimization technique like regression, PSO (Particle Swamp direct liquefaction are highly aromatic. These chemical Optimization) validating it with experimental values. The differences affect the end use. Indirect liquefaction provides validating incorporate kinetic rate constant, conversion to cross high-quality diesel fuel and intermediates for olefin verify the result obtained through optimization. production; direct liquefaction yields high-octane gasoline and Scope of this work is to get a better insight of coal excellent feedstocks for aromatic chemicals. -

Friedrich Bergius and the Rise of the German Synthetic Fuel Industry
Friedrich Bergius and the Rise of the German Synthetic Fue nI ustry By Anthony N. Stranges* G ERMANY HAS VIRTUALLY NO petroleumdeposits. Prior to the twen- tieth century that lack of a liquid fuel was not a serious problem, because Germany possessed abundantcoal reserves. Coal provided for commercial and home heating; it also fulfilled the needs of industryand the military,particularly the navy. In the first decade of the twentieth century, Germany's energy requirements began to change. Two reasons were especially important. First, Germany be- came increasinglydependent on gasoline and diesel oil engines. The appearance of automobiles, trucks, and then airplanes made a plentiful supply of gasoline absolutely essential; moreover, ocean-going ships increasingly used diesel oil rather than coal as their energy source. Second, Germany's continuing indus- trialization and urbanizationmagnified the shortcomings of coal as an energy source. German scientists and engineers began to replace coal with smokeless liquid fuels, which not only were cleaner burningand more convenient to handle but also had a higher energy content. Petroleumwas clearly the fuel of the future, and to insure that Germanywould never be without it, her scientists and engineers created a domestic source of that fuel. From a plentiful natural substance, coal, they synthesized petroleum. Of the several processes the Germansused to convert coal into petroleum, high- pressure coal hydrogenationwas the most highly advanced. Its history falls into two broad periods: 1910-1925, duringwhich time its inventor, FriedrichBergius (1884-1949) developed the process through the first stages of industrialization, and 1925-1945, the period of its further commercial development by German industrialists. -
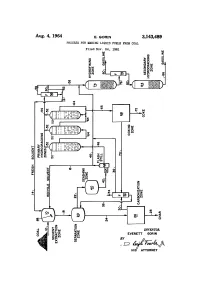
Aug. 4, 1964 E. Gorln 3,143,489 PROCESS for MAKING LIQUID FUELS from COAL Filed NOV
Aug. 4, 1964 E. GoRlN 3,143,489 PROCESS FOR MAKING LIQUID FUELS FROM COAL Filed NOV. 24, 1961 Immun I.. 3,143,489 United States Patent C) ice Patented Aug. d, i‘àfîlâ 1 2 For example, in the Pott-Broche Process as close to 100 3,143,489 weight percent of the coal as possible is converted to coal PRÜCESS FÜR MAKING ‘LIQUID FUELS extract. It was believed the resulting coal extract would FROM CGAL Everett Gerin, Pittsburgh, Pa., assigner to Consolidation be more amenable to subsequent catalytic hydrogenation Coal Company, Pittsburgh, Pa., a corporation of 5 than the original coal. Unfortunately, when substantial Pennsylvania ly all of the coal is converted to extract, the resulting ex Filed Nov. 24, 1961, Ser. No. 154,451 tract is as diflicult to hydrogenate as the coal. More 9 Claims. (Cl. 208-8) over, the expense of extracting substantially all of the coal is economically prohibitive, as fully discussed in my This invention relates lto a process for making liquid copending application, Serial No. 61,518, filed October fuels from coal. More particularly, this invention relates l0, 1960, now Patent No. 3,018,242 which is assigned to to a process for making gasoline from coal at a cost the assignee of the kpresent application. which is equal to or less than the cost per gallon of simi I have found that liquid fuels such as gasoline can be lar gasoline made from petroleum (based on present economically obtained from coal if the coal is subjected coal and petroleum prices). to a “partial conversion process.” By this I mean a proc For many years investigators have been trying to de ess wherein the coal and the intermediate coal-derived velop commercially feasible processes for the production products are sequentially and incrementally upgraded in of hydrogen-enriched liquid fuels such as gasoline from a series of process steps, each of which is designed to par materials other than petroleum. -

Hydrocarbon Liquefaction: Viability As a Peak Oil Mitigation Strategy
Munich Personal RePEc Archive Hydrocarbon liquefaction: viability as a peak oil mitigation strategy Höök, Mikael and Fantazzini, Dean and Angelantoni, André and Snowden, Simon Global Energy Systems, Department of Earth Sciences, Uppsala University (Sweden), Moscow School of Economics, Moscow State University, Moscow (Russia), Post Peak Living, San Francisco, USA„ Management School, University of Liverpool, United Kingdom 2013 Online at https://mpra.ub.uni-muenchen.de/46957/ MPRA Paper No. 46957, posted 13 May 2013 21:03 UTC Accepted by Philosophical Transactions of the Royal Society: A Hydrocarbon liquefaction: viability as a peak oil mitigation strategy Mikael Höök1, Dean Fantazzini234, André Angelantoni5, Simon Snowden6 Contact e-mail: [email protected] 1 Global Energy Systems, Department of Earth Sciences, Uppsala University, Villavägen 16, 752 36 Uppsala, Sweden, Telephone: +46 18-4713777, Fax: +46 184713513, web: http://www.geo.uu.se 2 Moscow School of Economics, Moscow State University, Moscow, Russia, www.mse-msu.ru 3 Faculty of Economics, Higher School of Economics, Moscow, Russia 4 The International College of Economics and Finance, Higher School of Economics, Moscow, Russia 5 Post Peak Living, San Francisco, USA, http://www.PostPeakLiving.com 6 Management School, University of Liverpool, United Kingdom Abstract Current world capacity of hydrocarbon liquefaction is around 400,000 barrels per day (kb/d), providing a marginal share of the global liquid fuel supply. This study performs a broad review of technical, economic, environmental, and supply chains issues related to coal-to- liquids (CTL) and gas-to-liquids (GTL). We find three issues predominate. First, significant amounts of coal and gas would be required to obtain anything more than a marginal production of liquids. -
Presentation 80A Stranges Germany
GERMANY’S SYNTHETIC FUEL INDUSTRIES Anthony Stranges Texas A&M University Department of History Abstract Germany’s Synthetic Fuel Industry 1930-1945 Abstract Petroleum was clearly the fuel of the future, and to insure that Germany would never lack a plentiful supply, German scientists and engineers synthesized petroleum from their country’s abundant coal supplies and thereby established the world’s first technologically successful synthetic fuels industry. Of the several conversion processes the Germans developed, high-pressure coal hydrogenation or liquefaction and the Fischer-Tropsch synthesis (F-T) were the most advanced. Friedrich Bergius (1884- 1949) launched the German program for energy independence with the invention of high-pressure coal hydrogenation in the years 1910-25. Franz Fischer (1877-1947) and Hans Tropsch broadened it with the gaseous synthesis of liquid fuels in the mid-1920s. IG Farben, Ruhrchemie, and others industrialized the German energy program with their development of the Bergius process and Fischer-Tropsch synthesis from the 1920s to the end of World War II. This paper examines Germany’s industrialization of the two synthetic fuel processes, analyzing their growth over several decades of social, political, and economic unrest. Overview SYNTHETIC FUELS IN GERMANY OVERVIEW 1911-1945 For the Fischer-Tropsch process the research and development falls into two divisions PERIOD ACTIVITY 1922-1935 Laboratory research and pilot plant development 1936-1945 Industrial-size development, with most of the development occurring -

Aviation Gasoline Technology Transfer During the Second World War: Japan, Germany and the U.S.A
Aviation Gasoline Technology Transfer during the Second World War: Japan, Germany and the U.S.A. Munehiro Miwa INTRODUCTION As an example of the reception of knowledge, let us consider the aviation gasoline production technologies, which were transferred over the boundaries of Allies vs. Axis Powers during the World War II. It is worthwhile to trace how Japan acquired the overseas methods from a web of newest and complicated technological information then available and developed its own technology. Detailed consideration reveals prejudiced assumptions, misleading reception of knowledge, technology transfer from enemy country, and other complex aspects. Why did Japan, specifically the Imperial Navy, believe that Germany succeeded in coal liquefaction by Bergius Process to produce aviation gasoline, and did not consider using low- temperature coal tar as the raw material? It is necessary to discuss the background and the reasons how the Naval Fuel Depot, which employed the most advanced technical staff in Japan at the time, led this move on the wrong assumption. The Navy doggedly insisted on the liquefaction process, spent enormous resources, and solicited the South Manchuria Railway Co., Ltd. (Fushun) and Nippon Chisso (Hungnam) to prepare for manufacture. The Navy showed interest in the information regarding the isooctane process, which came from the Universal Oil Products Company (“UOP”), and was actually able to show some results in production. However, it paid no heed to the similar, more efficient alkylation process, which played an important role in increased production of high-octane aviation spirit in the U.S. , in the Trinidad, in Abadan and in Germany during the war.