THE BINARY COLLISION APPROXIMATION: Iliiilms BACKGROUND and INTRODUCTION Mark T
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Astrodynamics
Politecnico di Torino SEEDS SpacE Exploration and Development Systems Astrodynamics II Edition 2006 - 07 - Ver. 2.0.1 Author: Guido Colasurdo Dipartimento di Energetica Teacher: Giulio Avanzini Dipartimento di Ingegneria Aeronautica e Spaziale e-mail: [email protected] Contents 1 Two–Body Orbital Mechanics 1 1.1 BirthofAstrodynamics: Kepler’sLaws. ......... 1 1.2 Newton’sLawsofMotion ............................ ... 2 1.3 Newton’s Law of Universal Gravitation . ......... 3 1.4 The n–BodyProblem ................................. 4 1.5 Equation of Motion in the Two-Body Problem . ....... 5 1.6 PotentialEnergy ................................. ... 6 1.7 ConstantsoftheMotion . .. .. .. .. .. .. .. .. .... 7 1.8 TrajectoryEquation .............................. .... 8 1.9 ConicSections ................................... 8 1.10 Relating Energy and Semi-major Axis . ........ 9 2 Two-Dimensional Analysis of Motion 11 2.1 ReferenceFrames................................. 11 2.2 Velocity and acceleration components . ......... 12 2.3 First-Order Scalar Equations of Motion . ......... 12 2.4 PerifocalReferenceFrame . ...... 13 2.5 FlightPathAngle ................................. 14 2.6 EllipticalOrbits................................ ..... 15 2.6.1 Geometry of an Elliptical Orbit . ..... 15 2.6.2 Period of an Elliptical Orbit . ..... 16 2.7 Time–of–Flight on the Elliptical Orbit . .......... 16 2.8 Extensiontohyperbolaandparabola. ........ 18 2.9 Circular and Escape Velocity, Hyperbolic Excess Speed . .............. 18 2.10 CosmicVelocities -

A Hot Subdwarf-White Dwarf Super-Chandrasekhar Candidate
A hot subdwarf–white dwarf super-Chandrasekhar candidate supernova Ia progenitor Ingrid Pelisoli1,2*, P. Neunteufel3, S. Geier1, T. Kupfer4,5, U. Heber6, A. Irrgang6, D. Schneider6, A. Bastian1, J. van Roestel7, V. Schaffenroth1, and B. N. Barlow8 1Institut fur¨ Physik und Astronomie, Universitat¨ Potsdam, Haus 28, Karl-Liebknecht-Str. 24/25, D-14476 Potsdam-Golm, Germany 2Department of Physics, University of Warwick, Coventry, CV4 7AL, UK 3Max Planck Institut fur¨ Astrophysik, Karl-Schwarzschild-Straße 1, 85748 Garching bei Munchen¨ 4Kavli Institute for Theoretical Physics, University of California, Santa Barbara, CA 93106, USA 5Texas Tech University, Department of Physics & Astronomy, Box 41051, 79409, Lubbock, TX, USA 6Dr. Karl Remeis-Observatory & ECAP, Astronomical Institute, Friedrich-Alexander University Erlangen-Nuremberg (FAU), Sternwartstr. 7, 96049 Bamberg, Germany 7Division of Physics, Mathematics and Astronomy, California Institute of Technology, Pasadena, CA 91125, USA 8Department of Physics and Astronomy, High Point University, High Point, NC 27268, USA *[email protected] ABSTRACT Supernova Ia are bright explosive events that can be used to estimate cosmological distances, allowing us to study the expansion of the Universe. They are understood to result from a thermonuclear detonation in a white dwarf that formed from the exhausted core of a star more massive than the Sun. However, the possible progenitor channels leading to an explosion are a long-standing debate, limiting the precision and accuracy of supernova Ia as distance indicators. Here we present HD 265435, a binary system with an orbital period of less than a hundred minutes, consisting of a white dwarf and a hot subdwarf — a stripped core-helium burning star. -

Electric Propulsion System Scaling for Asteroid Capture-And-Return Missions
Electric propulsion system scaling for asteroid capture-and-return missions Justin M. Little⇤ and Edgar Y. Choueiri† Electric Propulsion and Plasma Dynamics Laboratory, Princeton University, Princeton, NJ, 08544 The requirements for an electric propulsion system needed to maximize the return mass of asteroid capture-and-return (ACR) missions are investigated in detail. An analytical model is presented for the mission time and mass balance of an ACR mission based on the propellant requirements of each mission phase. Edelbaum’s approximation is used for the Earth-escape phase. The asteroid rendezvous and return phases of the mission are modeled as a low-thrust optimal control problem with a lunar assist. The numerical solution to this problem is used to derive scaling laws for the propellant requirements based on the maneuver time, asteroid orbit, and propulsion system parameters. Constraining the rendezvous and return phases by the synodic period of the target asteroid, a semi- empirical equation is obtained for the optimum specific impulse and power supply. It was found analytically that the optimum power supply is one such that the mass of the propulsion system and power supply are approximately equal to the total mass of propellant used during the entire mission. Finally, it is shown that ACR missions, in general, are optimized using propulsion systems capable of processing 100 kW – 1 MW of power with specific impulses in the range 5,000 – 10,000 s, and have the potential to return asteroids on the order of 103 104 tons. − Nomenclature -
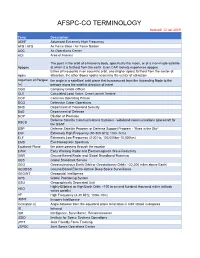
AFSPC-CO TERMINOLOGY Revised: 12 Jan 2019
AFSPC-CO TERMINOLOGY Revised: 12 Jan 2019 Term Description AEHF Advanced Extremely High Frequency AFB / AFS Air Force Base / Air Force Station AOC Air Operations Center AOI Area of Interest The point in the orbit of a heavenly body, specifically the moon, or of a man-made satellite Apogee at which it is farthest from the earth. Even CAP rockets experience apogee. Either of two points in an eccentric orbit, one (higher apsis) farthest from the center of Apsis attraction, the other (lower apsis) nearest to the center of attraction Argument of Perigee the angle in a satellites' orbit plane that is measured from the Ascending Node to the (ω) perigee along the satellite direction of travel CGO Company Grade Officer CLV Calculated Load Value, Crew Launch Vehicle COP Common Operating Picture DCO Defensive Cyber Operations DHS Department of Homeland Security DoD Department of Defense DOP Dilution of Precision Defense Satellite Communications Systems - wideband communications spacecraft for DSCS the USAF DSP Defense Satellite Program or Defense Support Program - "Eyes in the Sky" EHF Extremely High Frequency (30-300 GHz; 1mm-1cm) ELF Extremely Low Frequency (3-30 Hz; 100,000km-10,000km) EMS Electromagnetic Spectrum Equitorial Plane the plane passing through the equator EWR Early Warning Radar and Electromagnetic Wave Resistivity GBR Ground-Based Radar and Global Broadband Roaming GBS Global Broadcast Service GEO Geosynchronous Earth Orbit or Geostationary Orbit ( ~22,300 miles above Earth) GEODSS Ground-Based Electro-Optical Deep Space Surveillance -

Use of MESSENGER Radioscience Data to Improve Planetary Ephemeris and to Test General Relativity
A&A 561, A115 (2014) Astronomy DOI: 10.1051/0004-6361/201322124 & © ESO 2014 Astrophysics Use of MESSENGER radioscience data to improve planetary ephemeris and to test general relativity A. K. Verma1;2, A. Fienga3;4, J. Laskar4, H. Manche4, and M. Gastineau4 1 Observatoire de Besançon, UTINAM-CNRS UMR6213, 41bis avenue de l’Observatoire, 25000 Besançon, France e-mail: [email protected] 2 Centre National d’Études Spatiales, 18 avenue Édouard Belin, 31400 Toulouse, France 3 Observatoire de la Côte d’Azur, GéoAzur-CNRS UMR7329, 250 avenue Albert Einstein, 06560 Valbonne, France 4 Astronomie et Systèmes Dynamiques, IMCCE-CNRS UMR8028, 77 Av. Denfert-Rochereau, 75014 Paris, France Received 24 June 2013 / Accepted 7 November 2013 ABSTRACT The current knowledge of Mercury’s orbit has mainly been gained by direct radar ranging obtained from the 60s to 1998 and by five Mercury flybys made with Mariner 10 in the 70s, and with MESSENGER made in 2008 and 2009. On March 18, 2011, MESSENGER became the first spacecraft to orbit Mercury. The radioscience observations acquired during the orbital phase of MESSENGER drastically improved our knowledge of the orbit of Mercury. An accurate MESSENGER orbit is obtained by fitting one-and-half years of tracking data using GINS orbit determination software. The systematic error in the Earth-Mercury geometric positions, also called range bias, obtained from GINS are then used to fit the INPOP dynamical modeling of the planet motions. An improved ephemeris of the planets is then obtained, INPOP13a, and used to perform general relativity tests of the parametrized post-Newtonian (PPN) formalism. -

Multi-Body Trajectory Design Strategies Based on Periapsis Poincaré Maps
MULTI-BODY TRAJECTORY DESIGN STRATEGIES BASED ON PERIAPSIS POINCARÉ MAPS A Dissertation Submitted to the Faculty of Purdue University by Diane Elizabeth Craig Davis In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy August 2011 Purdue University West Lafayette, Indiana ii To my husband and children iii ACKNOWLEDGMENTS I would like to thank my advisor, Professor Kathleen Howell, for her support and guidance. She has been an invaluable source of knowledge and ideas throughout my studies at Purdue, and I have truly enjoyed our collaborations. She is an inspiration to me. I appreciate the insight and support from my committee members, Professor James Longuski, Professor Martin Corless, and Professor Daniel DeLaurentis. I would like to thank the members of my research group, past and present, for their friendship and collaboration, including Geoff Wawrzyniak, Chris Patterson, Lindsay Millard, Dan Grebow, Marty Ozimek, Lucia Irrgang, Masaki Kakoi, Raoul Rausch, Matt Vavrina, Todd Brown, Amanda Haapala, Cody Short, Mar Vaquero, Tom Pavlak, Wayne Schlei, Aurelie Heritier, Amanda Knutson, and Jeff Stuart. I thank my parents, David and Jeanne Craig, for their encouragement and love throughout my academic career. They have cheered me on through many years of studies. I am grateful for the love and encouragement of my husband, Jonathan. His never-ending patience and friendship have been a constant source of support. Finally, I owe thanks to the organizations that have provided the funding opportunities that have supported me through my studies, including the Clare Booth Luce Foundation, Zonta International, and Purdue University and the School of Aeronautics and Astronautics through the Graduate Assistance in Areas of National Need and the Purdue Forever Fellowships. -

Perturbation Theory in Celestial Mechanics
Perturbation Theory in Celestial Mechanics Alessandra Celletti Dipartimento di Matematica Universit`adi Roma Tor Vergata Via della Ricerca Scientifica 1, I-00133 Roma (Italy) ([email protected]) December 8, 2007 Contents 1 Glossary 2 2 Definition 2 3 Introduction 2 4 Classical perturbation theory 4 4.1 The classical theory . 4 4.2 The precession of the perihelion of Mercury . 6 4.2.1 Delaunay action–angle variables . 6 4.2.2 The restricted, planar, circular, three–body problem . 7 4.2.3 Expansion of the perturbing function . 7 4.2.4 Computation of the precession of the perihelion . 8 5 Resonant perturbation theory 9 5.1 The resonant theory . 9 5.2 Three–body resonance . 10 5.3 Degenerate perturbation theory . 11 5.4 The precession of the equinoxes . 12 6 Invariant tori 14 6.1 Invariant KAM surfaces . 14 6.2 Rotational tori for the spin–orbit problem . 15 6.3 Librational tori for the spin–orbit problem . 16 6.4 Rotational tori for the restricted three–body problem . 17 6.5 Planetary problem . 18 7 Periodic orbits 18 7.1 Construction of periodic orbits . 18 7.2 The libration in longitude of the Moon . 20 1 8 Future directions 20 9 Bibliography 21 9.1 Books and Reviews . 21 9.2 Primary Literature . 22 1 Glossary KAM theory: it provides the persistence of quasi–periodic motions under a small perturbation of an integrable system. KAM theory can be applied under quite general assumptions, i.e. a non– degeneracy of the integrable system and a diophantine condition of the frequency of motion. -

On Optimal Two-Impulse Earth–Moon Transfers in a Four-Body Model
Noname manuscript No. (will be inserted by the editor) On Optimal Two-Impulse Earth–Moon Transfers in a Four-Body Model F. Topputo Received: date / Accepted: date Abstract In this paper two-impulse Earth–Moon transfers are treated in the restricted four-body problem with the Sun, the Earth, and the Moon as primaries. The problem is formulated with mathematical means and solved through direct transcription and multiple shooting strategy. Thousands of solutions are found, which make it possible to frame known cases as special points of a more general picture. Families of solutions are defined and characterized, and their features are discussed. The methodology described in this paper is useful to perform trade-off analyses, where many solutions have to be produced and assessed. Keywords Earth–Moon transfer low-energy transfer ballistic capture trajectory optimization · · · · restricted three-body problem restricted four-body problem · 1 Introduction The search for trajectories to transfer a spacecraft from the Earth to the Moon has been the subject of countless works. The Hohmann transfer represents the easiest way to perform an Earth–Moon transfer. This requires placing the spacecraft on an ellipse having the perigee on the Earth parking orbit and the apogee on the Moon orbit. By properly switching the gravitational attractions along the orbit, the spacecraft’s motion is governed by only the Earth for most of the transfer, and by only the Moon in the final part. More generally, the patched-conics approximation relies on a Keplerian decomposition of the solar system dynamics (Battin 1987). Although from a practical point of view it is desirable to deal with analytical solutions, the two-body problem being integrable, the patched-conics approximation inherently involves hyperbolic approaches upon arrival. -

Wobbling Stars and Planets in This Activity, You
Activity: Wobbling Stars and Planets In this activity, you will investigate the effect of the relative masses and distances of objects that are in orbit around a star or planet. The most common understanding of how the planets orbit around the Sun is that the Sun is stationary while the planets revolve around the Sun. That is not quite true and is a little more complicated. PBS An important parameter when considering orbits is the center of mass. The concept of the center of mass is that of an average of the masses factored by their distances from one another. This is also commonly referred to as the center of gravity. For example, the balancing of a ETIC seesaw about a pivot point demonstrates the center of mass of two objects of different masses. Another important parameter to consider is the barycenter. The barycenter is the center of mass of two or more bodies that are orbiting each other, and is the point around which both of them orbit. When the two bodies have similar masses, the barycenter may be located between NASA the two bodies (outside of either of bodies) and both objects will follow an orbit around the center of mass. This is the case for a system such as the Sun and Jupiter. In the case where the two objects have very different masses, the center of mass and barycenter may be located within the more massive body. This is the case for the Earth-Moon system. scienceprojectideasforkids.com Classroom Activity Part 1 Materials • Wooden skewers 1. -

Exploring the Orbital Evolution of Planetary Systems
Exploring the orbital evolution of planetary systems Tabar´e Gallardo∗ Instituto de F´ısica, Facultad de Ciencias, Udelar. (Dated: January 22, 2017) The aim of this paper is to encourage the use of orbital integrators in the classroom to discover and understand the long term dynamical evolution of systems of orbiting bodies. We show how to perform numerical simulations and how to handle the output data in order to reveal the dynamical mechanisms that dominate the evolution of arbitrary planetary systems in timescales of millions of years using a simple but efficient numerical integrator. Trough some examples we reveal the fundamental properties of the planetary systems: the time evolution of the orbital elements, the free and forced modes that drive the oscillations in eccentricity and inclination, the fundamental frequencies of the system, the role of the angular momenta, the invariable plane, orbital resonances and the Kozai-Lidov mechanism. To be published in European Journal of Physics. 2 I. INTRODUCTION With few exceptions astronomers cannot make experiments, they are limited to observe the universe. The laboratory for the astronomer usually takes the form of computer simulations. This is the most important instrument for the study of the dynamical behavior of gravitationally interacting bodies. A planetary system, for example, evolves mostly due to gravitation acting over very long timescales generating what is called a secular evolution. This secular evolution can be deduced analytically by means of the theory of perturbations, but can also be explored in the classroom by means of precise numerical integrators. Some facilities exist to visualize and experiment with the gravitational interactions between massive bodies1–3. -

A Delta-V Map of the Known Main Belt Asteroids
Acta Astronautica 146 (2018) 73–82 Contents lists available at ScienceDirect Acta Astronautica journal homepage: www.elsevier.com/locate/actaastro A Delta-V map of the known Main Belt Asteroids Anthony Taylor *, Jonathan C. McDowell, Martin Elvis Harvard-Smithsonian Center for Astrophysics, 60 Garden St., Cambridge, MA, USA ARTICLE INFO ABSTRACT Keywords: With the lowered costs of rocket technology and the commercialization of the space industry, asteroid mining is Asteroid mining becoming both feasible and potentially profitable. Although the first targets for mining will be the most accessible Main belt near Earth objects (NEOs), the Main Belt contains 106 times more material by mass. The large scale expansion of NEO this new asteroid mining industry is contingent on being able to rendezvous with Main Belt asteroids (MBAs), and Delta-v so on the velocity change required of mining spacecraft (delta-v). This paper develops two different flight burn Shoemaker-Helin schemes, both starting from Low Earth Orbit (LEO) and ending with a successful MBA rendezvous. These methods are then applied to the 700,000 asteroids in the Minor Planet Center (MPC) database with well-determined orbits to find low delta-v mining targets among the MBAs. There are 3986 potential MBA targets with a delta- v < 8kmsÀ1, but the distribution is steep and reduces to just 4 with delta-v < 7kms-1. The two burn methods are compared and the orbital parameters of low delta-v MBAs are explored. 1. Introduction [2]. A running tabulation of the accessibility of NEOs is maintained on- line by Lance Benner3 and identifies 65 NEOs with delta-v< 4:5kms-1.A For decades, asteroid mining and exploration has been largely dis- separate database based on intensive numerical trajectory calculations is missed as infeasible and unprofitable. -

Orbital Mechanics
Orbital Mechanics Part 1 Orbital Forces Why a Sat. remains in orbit ? Bcs the centrifugal force caused by the Sat. rotation around earth is counter- balanced by the Earth's Pull. Kepler’s Laws The Satellite (Spacecraft) which orbits the earth follows the same laws that govern the motion of the planets around the sun. J. Kepler (1571-1630) was able to derive empirically three laws describing planetary motion I. Newton was able to derive Keplers laws from his own laws of mechanics [gravitation theory] Kepler’s 1st Law (Law of Orbits) The path followed by a Sat. (secondary body) orbiting around the primary body will be an ellipse. The center of mass (barycenter) of a two-body system is always centered on one of the foci (earth center). Kepler’s 1st Law (Law of Orbits) The eccentricity (abnormality) e: a 2 b2 e a b- semiminor axis , a- semimajor axis VIN: e=0 circular orbit 0<e<1 ellip. orbit Orbit Calculations Ellipse is the curve traced by a point moving in a plane such that the sum of its distances from the foci is constant. Kepler’s 2nd Law (Law of Areas) For equal time intervals, a Sat. will sweep out equal areas in its orbital plane, focused at the barycenter VIN: S1>S2 at t1=t2 V1>V2 Max(V) at Perigee & Min(V) at Apogee Kepler’s 3rd Law (Harmonic Law) The square of the periodic time of orbit is proportional to the cube of the mean distance between the two bodies. a 3 n 2 n- mean motion of Sat.