Crystals with Disordered Hydrogen-Bond Networks and Superprotonic Conductivity
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
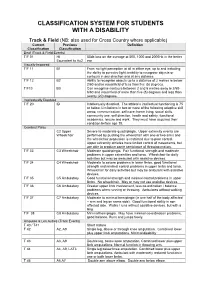
Disability Classification System
CLASSIFICATION SYSTEM FOR STUDENTS WITH A DISABILITY Track & Field (NB: also used for Cross Country where applicable) Current Previous Definition Classification Classification Deaf (Track & Field Events) T/F 01 HI 55db loss on the average at 500, 1000 and 2000Hz in the better Equivalent to Au2 ear Visually Impaired T/F 11 B1 From no light perception at all in either eye, up to and including the ability to perceive light; inability to recognise objects or contours in any direction and at any distance. T/F 12 B2 Ability to recognise objects up to a distance of 2 metres ie below 2/60 and/or visual field of less than five (5) degrees. T/F13 B3 Can recognise contours between 2 and 6 metres away ie 2/60- 6/60 and visual field of more than five (5) degrees and less than twenty (20) degrees. Intellectually Disabled T/F 20 ID Intellectually disabled. The athlete’s intellectual functioning is 75 or below. Limitations in two or more of the following adaptive skill areas; communication, self-care; home living, social skills, community use, self direction, health and safety, functional academics, leisure and work. They must have acquired their condition before age 18. Cerebral Palsy C2 Upper Severe to moderate quadriplegia. Upper extremity events are Wheelchair performed by pushing the wheelchair with one or two arms and the wheelchair propulsion is restricted due to poor control. Upper extremity athletes have limited control of movements, but are able to produce some semblance of throwing motion. T/F 33 C3 Wheelchair Moderate quadriplegia. Fair functional strength and moderate problems in upper extremities and torso. -

Sapphire: Properties, Growth, and Applications
Sapphire: Properties, Growth, and 1904), which is sometimes called the flame fusion method. In this technique, alumina powder to be Applications crystallized is fused in a hydrogen–oxygen flame and falls on the molten upper surface of an oriented seed Sapphire has a high refractive index and a broad crystal placed in a furnace (Fig. 1(a)). transmission band spanning the UV, visible, and IR Since this is not a refined process, the material can bands. Sapphire also has a high hardness and melting contain voids and powder inclusions and is likely to be point, and very good thermal conductivity, tensile strained. However, crystals of up to 60mm in diameter strength, and thermal shock resistance (Table 1). The can be grown by this method. Figure 2 shows typical favorable combination of excellent optical and mech- sapphire boules grown by the Verneuil technique. anical properties of sapphire, together with high chemical durability, makes it an attractive structural material for high-technology applications. Sapphire crystals are used in medicine and blood chemistry as 1.2 Czochralski Growth they are resistant to human blood and body fluids, and are totally impervious to moisture and chemically This technique originates from pioneering work by inert. Frequently it is the combination of two, or more, Czochralski (1917). Crystals grow from the meniscus of its properties that make sapphire the only material formed on a free surface of the melt onto the seed available to solve complex engineering design crystal of the required crystallographic orientation. problems. The pull rod is lifted and rotated, and crystallization However, sapphire is difficult to shape owing to its onto the end of the seed occurs (Fig. -

Mineral Processing
Mineral Processing Foundations of theory and practice of minerallurgy 1st English edition JAN DRZYMALA, C. Eng., Ph.D., D.Sc. Member of the Polish Mineral Processing Society Wroclaw University of Technology 2007 Translation: J. Drzymala, A. Swatek Reviewer: A. Luszczkiewicz Published as supplied by the author ©Copyright by Jan Drzymala, Wroclaw 2007 Computer typesetting: Danuta Szyszka Cover design: Danuta Szyszka Cover photo: Sebastian Bożek Oficyna Wydawnicza Politechniki Wrocławskiej Wybrzeze Wyspianskiego 27 50-370 Wroclaw Any part of this publication can be used in any form by any means provided that the usage is acknowledged by the citation: Drzymala, J., Mineral Processing, Foundations of theory and practice of minerallurgy, Oficyna Wydawnicza PWr., 2007, www.ig.pwr.wroc.pl/minproc ISBN 978-83-7493-362-9 Contents Introduction ....................................................................................................................9 Part I Introduction to mineral processing .....................................................................13 1. From the Big Bang to mineral processing................................................................14 1.1. The formation of matter ...................................................................................14 1.2. Elementary particles.........................................................................................16 1.3. Molecules .........................................................................................................18 1.4. Solids................................................................................................................19 -
Crystal Growth and Defects
Hartmut S. Leipner, Reinhard Krause-Rehberg Structure of imperfect solids (2) A. Advanced topics B. Methods Syllabus 1–2. Defects and crystal growth 3–4. Defects and semiconductor technology 5–7.* Defect engineering; diffusion 8. Optical methods 9. * Electrical methods 10. * Positron annihilation 11. * Resonance techniques 12. X-ray methods 13. * Probe techniques (* given by Reinhard Krause-Rehberg) 2 Summary As the continuation of the introduction into crystal defects (in the SS 2001), advanced topics of solid state physics related to defects are treated in this semester. Topics are the crystal growth from the point of view of crystal imperfections, diffusion in solids, and the role of defects in the production and the function of solid state devices. In the second part of this lecture, basic experimental techniques of the investigation of defects are introduced. The following methods are treated: optical and electrical methods (luminescence, Hall effect, DLTS), X-ray techniques, probe and resonance techniques (positron annihilation, pertubed angular correlation, electron paramagnetic resonance). The pieces of information to be extracted from the particular methods for the characterization of the defect structure are discussed. 3 Literature K.-T. Wilke: Kristallzüchtung. Berlin: Deutscher Verlag der Wissenschaften 1988. Silicon devices. Ed. K. A. Jackson. Weinheim: Wiley-VCH 1998. Bergmann Schäfer Lehrbuch der Experimentalphysik. Band 6 Festkörper. Hrg. W. Raith. Berlin: De Gruyter 1992. B. G. Jacobi, D. B. Holt: Cathodoluminescence microscopy of inorganic solids. New York: Plenum 1990. S. Pfüller: Halbleitermeßtechnik. Berlin: Verlag Technik 1976. G. Schatz, A. Weidinger: Nukleare Festkörperphysik. Stuttgart: Teubner 1992. Identification of defects in semiconductors. Ed. M. Stavola. San Diego: Academic Press 1998 Hartmut S. -

A Mineralogy of Anthropocene E
1 A Minerology for the Anthropocene Pierre FLUCK Institut Universitaire de France / Docteur-ès-Sciences / geologist and archeologist / Emeritus Professor at Université de Haute-Alsace This essay is a follow-up on « La signature stratigraphique de l’Anthropocène », which is also available on HAL- Archives ouvertes. Table of contents 1. Introduction: neoformation minerals in ancient mining galleries 2. Minerals from burning coal mines 3. Minerals from the mineral processing industry 4 ...and metallurgy 5. Neoformations in slags 6. Speciation of heavy metals in soils 7. Metal objects in their archaeological environment, or affected by fire 8. Neoformations in or on the surface of building stones 9. A mineralogy of materials. The “miracle of the potter”. The minerals in cement 10. A mineralogy of the biosphere? Conclusions Warning. This paper is written to be read by both specialists and a wider audience. However, it contains many mineral names. While these may resonate in the minds of mineralogists or collectors, they may not be as meaningful to less discerning readers. Such readers should not be scared, for they may find excellent encyclopaedic records on the web, including chemical composition, crystallographic properties and description of each of these species. This is why we have decided not to include further information in this paper. Acknowledgements. I would like to thank the mineralogists with whom I have had the opportunity to maintain fruitful exchanges for a long time: my pupil Hubert Bari, Éric Asselborn, Cédric Lheur, François Farges. And I would like to honour the memories of René Weil (1901-1983), my master in descriptive mineralogy, and of Jacques Geffroy (1918-1993), pupil of Alfred Lacroix, my master in metallogeny. -

Development Team
Material science Paper No. : Crystallography & crystal growth Module : Growth from melt II Development Team Prof. Vinay Gupta, Department of Physics and Astrophysics, Principal Investigator University of Delhi, Delhi Prof. P. N. Kotru ,Department of Physics, University of Jammu, Paper Coordinator Jammu-180006 Content Writer Prof. P. N. Kotru ,Department of Physics, University of Jammu, Jammu-180006 Prof Mahavir Singh Department of Physics, Himachal Pradesh Content Reviewer University, Shimla 1 Crystallography & crystal growth Material science Growth from melt II Description of Module Subject Name Physics Paper Name Crystallography & crystal growth Module Name/Title Growth from melt II Module Id 31 2 Crystallography & crystal growth Material science Growth from melt II 31 Bridgman-Stockbarger Growth Technique. 31.1 Introduction The techniques were originated by Bridgman (1925) and Stockbarger (1938) and so are named after them. In these techniques a crucible containing the material to be grown is lowered through a furnace in such a way that the lowest point in the crucible and the solidification surface rises slowly up the crucible. It means that the melt contained in the crucible is progressively frozen to yield a single crystal. The rate of lowering the crucible may range from about 0.1 to 200 mmh─1 but in most of the cases it may range somewhere in between 1-30 mmh─1. There are situations where the movement of the crucible is reversed. In other words, the crucible is raised up through the furnace and so is advantageously applicable for materials which are volatile; the interface with the vapour being the coolest part of the charge. -

Ga2o3 and Its Vertical Structure Leds
micromachines Review Epitaxy of III-Nitrides on β-Ga2O3 and Its Vertical Structure LEDs Weijiang Li 1,2,3, Xiang Zhang 1,2,3, Ruilin Meng 1,2,3, Jianchang Yan 1,2,3, Junxi Wang 1,2,3, Jinmin Li 1,2,3 and Tongbo Wei 1,2,3,* 1 State Key Laboratory of Solid-State Lighting, Institute of Semiconductors, University of Chinese Academy of Sciences, Beijing 100083, China; [email protected] (W.L.); [email protected] (X.Z.); [email protected] (R.M.); [email protected] (J.Y.); [email protected] (J.W.); [email protected] (J.L.) 2 Center of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China 3 Beijing Engineering Research Center for the 3rd Generation Semiconductor Materials and Application, Beijing 100083, China * Correspondence: [email protected]; Tel.: +86-010-8230-5430 Received: 7 April 2019; Accepted: 8 May 2019; Published: 13 May 2019 Abstract: β-Ga2O3, characterized with high n-type conductivity, little lattice mismatch with III-Nitrides, high transparency (>80%) in blue, and UVA (400–320 nm) as well as UVB (320–280 nm) regions, has great potential as the substrate for vertical structure blue and especially ultra violet LEDs (light emitting diodes). Large efforts have been made to improve the quality of III-Nitrides epilayers on β-Ga2O3. Furthermore, the fabrication of vertical blue LEDs has been preliminarily realized with the best result that output power reaches to 4.82 W (under a current of 10 A) and internal quantum efficiency (IQE) exceeds 78% by different groups, respectively, while there is nearly no demonstration of UV-LEDs on β-Ga2O3. -

Villyaellenite (Mn, Ca)Mn2(Aso3oh)2(Aso4)2(H2O)4
Villyaellenite (Mn, Ca)Mn2(AsO3OH)2(AsO4)2(H2O)4 Crystal Data: Monoclinic. Point Group: 2/m. Crystals tabular on {100}, to prismatic along [001], showing {100}, {110}, {011}, {010}, {101}, and {001}, to 4 cm; in rosettes and radial aggregates. Physical Properties: Cleavage: Good on {100}. Hardness = ∼4 D(meas.) = 3.20-3.69 D(calc.) = 3.339 Optical Properties: Transparent. Color: Pale rose-red, orange-pink, colorless; colorless in transmitted light. Streak: White. Luster: Vitreous. Optical Class: Biaxial (-). Pleochroism: Moderate; X = very pale orange-pink; Y = exceedingly pale orange-pink; Z = pale orange-pink. Orientation: X = b; Y ∧ c = 30°-40°. Absorption: Z >> X > Y. α = 1.660-1.713 β = 1.670-1.723 γ = 1.676-1.729 2V(meas.) = 70.5°-76° 2V(calc.) = 75°-75.6° Cell Data: Space Group: C2/c. a = 18.400(2) b = 9.4778(10) c = 9.9594(12) β = 96.587(3)° Z = 4 X-ray Powder Pattern: Sainte-Marie-aux-Mines, France. 3.297 (100), 8.476 (90), 3.132 (60), 4.606 (50), 4.761 (40), 3.811 (40), 3.025 (40) Chemistry: (1) (2) As2O5 52.99 50.6 FeO 0.1 MnO 22.40 36.2 ZnO 2.9 CaO 13.58 0.5 H2O 11.42 9.9 Total 100.39 100.2 (1) Sainte-Marie-aux-Mines, France; by electron microprobe, total Mn as MnO, H2O by TGA; 2+ reducing H2O to 10.7% by analogy to other group members, corresponds to (Mn 2.74Ca2.10)Σ=4.84 (H2O)4(AsO3OH)2.31(AsO4)1.69. -

New Minerals Approved Bythe Ima Commission on New
NEW MINERALS APPROVED BY THE IMA COMMISSION ON NEW MINERALS AND MINERAL NAMES ALLABOGDANITE, (Fe,Ni)l Allabogdanite, a mineral dimorphous with barringerite, was discovered in the Onello iron meteorite (Ni-rich ataxite) found in 1997 in the alluvium of the Bol'shoy Dolguchan River, a tributary of the Onello River, Aldan River basin, South Yakutia (Republic of Sakha- Yakutia), Russia. The mineral occurs as light straw-yellow, with strong metallic luster, lamellar crystals up to 0.0 I x 0.1 x 0.4 rnrn, typically twinned, in plessite. Associated minerals are nickel phosphide, schreibersite, awaruite and graphite (Britvin e.a., 2002b). Name: in honour of Alia Nikolaevna BOG DAN OVA (1947-2004), Russian crys- tallographer, for her contribution to the study of new minerals; Geological Institute of Kola Science Center of Russian Academy of Sciences, Apatity. fMA No.: 2000-038. TS: PU 1/18632. ALLOCHALCOSELITE, Cu+Cu~+PbOZ(Se03)P5 Allochalcoselite was found in the fumarole products of the Second cinder cone, Northern Breakthrought of the Tolbachik Main Fracture Eruption (1975-1976), Tolbachik Volcano, Kamchatka, Russia. It occurs as transparent dark brown pris- matic crystals up to 0.1 mm long. Associated minerals are cotunnite, sofiite, ilin- skite, georgbokiite and burn site (Vergasova e.a., 2005). Name: for the chemical composition: presence of selenium and different oxidation states of copper, from the Greek aA.Ao~(different) and xaAxo~ (copper). fMA No.: 2004-025. TS: no reliable information. ALSAKHAROVITE-Zn, NaSrKZn(Ti,Nb)JSi401ZJz(0,OH)4·7HzO photo 1 Labuntsovite group Alsakharovite-Zn was discovered in the Pegmatite #45, Lepkhe-Nel'm MI. -

Structure of the Full Kinetoplastids Mitoribosome and Insight on Its Large Subunit Maturation
bioRxiv preprint doi: https://doi.org/10.1101/2020.05.02.073890; this version posted June 30, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Structure of the full kinetoplastids mitoribosome and insight on its large subunit maturation Heddy Soufari1* & Florent Waltz1*, Camila Parrot1+, Stéphanie Durrieu1+, Anthony Bochler1, Lauriane Kuhn2, Marie Sissler1, Yaser Hashem1‡ 1 Institut Européen de Chimie et Biologie, U1212 Inserm, UMR5320 CNRS, Université de Bordeaux, 2 rue R. Escarpit, F-33600 Pessac, France 2 Plateforme protéomique Strasbourg Esplanade FRC1589 du CNRS, Université de Strasbourg, Strasbourg, France *contributed equally, co-first authors +contributed equally, co-second authors ‡corresponding author Abstract: Kinetoplastids are unicellular eukaryotic parasites responsible for human pathologies such as Chagas disease, sleeping sickness or Leishmaniasis1. They possess a single large mitochondrion, essential for the parasite survival2. In kinetoplastids mitochondrion, most of the molecular machineries and gene expression processes have significantly diverged and specialized, with an extreme example being their mitochondrial ribosomes3. These large complexes are in charge of translating the few essential mRNAs encoded by mitochondrial genomes4,5. Structural studies performed in Trypanosoma brucei already highlighted the numerous peculiarities of these mitoribosomes and the maturation of their small subunit3,6. However, several important aspects mainly related to the large subunit remain elusive, such as the structure and maturation of its ribosomal RNA3. Here, we present a cryo-electron microscopy study of the protozoans Leishmania tarentolae and Trypanosoma cruzi mitoribosomes. -

RULES 2004 Final Amended
INTERNATIONAL PARALYMPIC COMMITTEE ATHLETICS SECTION COMBINED RULES 2004 PREAMBLE For competition at Paralympics and I.P.C. World Championships, this book shall be used, along with the current I.A.A.F. Handbook. It contains all the rules, which govern an I.P.C. Athletics competition, written in a way, which is compatible with the rules of the governing body for athletics, the International Association of Athletics Federations (I.A.A.F.). In this way, officials, coaches and athletes may find rules to cover any event in a single document, rather than having to refer to separate books for each group. The rules must be read in conjunction with the I.A.A.F. rules, contained in the Handbook of that Association. For the period including the 2004 Athens Paralympics, the version of the I.A.A.F. Handbook to which this book refers is the 2004-2005 edition. The rules concerned are the Technical Rules of Competition as described herein, and additional rules as shown. The reference to the I.A.A.F. Handbook does not confer any responsibility onto the I.A.A.F. for the I.P.C. Rules. Each of the I.O.S.D.’s has its own cycle of rule changes, and this volume does not intend to change any rules so decided. It is quite simply a means of simplifying the task of reading the rules for those concerned. NOTES This Rule Book will remain in force until the publication of the next I.A.A.F. Handbook, at which time a new edition will be published to take account of any new I.A.A.F. -

6H2O, a New Mineral and a Possible Sink for Sb During
1 Revision #1 2 Smamite, Ca2Sb(OH)4[H(AsO4)2]·6H2O, a new mineral and a possible sink for Sb during 3 weathering of fahlore 4 1§ 2 3 4 5 JAKUB PLÁŠIL , ANTHONY R. KAMPF , NICOLAS MEISSER , CÉDRIC LHEUR , THIERRY 5 6 6 BRUNSPERGER AND RADEK ŠKODA 7 8 1 Institute of Physics ASCR, v.v.i., Na Slovance 1999/2, 18221 Prague 8, Czech Republic 9 2 Mineral Sciences Department, Natural History Museum of Los Angeles County, 900 Exposition 10 Boulevard, Los Angeles, CA 90007, USA 11 3 Musée cantonal de géologie, Université de Lausanne, Anthropole, Dorigny, CH-1015 12 Lausanne, Switzerland 13 4 1 rue du St. Laurent, 54280 Seichamps, France 14 5 22 route de Wintzenheim, 68000 Colmar, France 15 6 Department of Geological Sciences, Faculty of Science, Masaryk University, Kotlářská 2, 611 16 37, Brno, Czech Republic 17 18 ABSTRACT 19 Smamite, Ca2Sb(OH)4[H(AsO4)2]·6H2O, is a new mineral species from the Giftgrube mine, 20 Rauenthal, Sainte-Marie-Aux-Mines ore-district, Haut-Rhin department, France. It is a supergene 21 mineral found in quartz-carbonate gangue with disseminated to massive tennantite-tetrahedrite 22 series minerals, native arsenic, Ni-Co arsenides and supergene minerals picropharmacolite, 23 fluckite and pharmacolite. Smamite occurs as lenticular crystals growing in aggregates up to 0.5 24 mm across. The new mineral is whitish to colorless, transparent with vitreous luster and white 25 streak; non-fluorescent under UV radiation. The Mohs hardness is ~3½ ; the tenacity is brittle, 26 the fracture is curved, and there is no apparent cleavage.