When Size Matters
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Softgels' Clear Advantages
DEEPDIVE REPORT March 2019 naturalproductsinsider.com Softgels’ Clear Advantages Report brought to you by DEEPDIVE REPORT Softgels’ Clear Advantages Contents Market snapshot ..................................................................................... 3 Consumer appeal .................................................................................... 4 Softgel history .......................................................................................... 6 Advantages of softgels ........................................................................... 7 How softgels are made .......................................................................... 9 Softgel challenges and solutions .......................................................11 Oxidation ...........................................................................................11 Consumer experience .......................................................................12 Dietary considerations ......................................................................12 Shelf stability ....................................................................................14 Bioavailability ....................................................................................14 Innovative developments .....................................................................16 Copyright © 2019 Informa Exhibitions LLC. All rights reserved. The publisher reserves the right to accept or reject any advertising or editorial material. Advertisers, and/or their agents, assume the -

Prescription Advantage Drug Exclusions
February 23, 2006 Dear Prescription Advantage Member: This letter is to inform you about changes to your Prescription Advantage coverage because of the new Medicare prescription drug coverage that began on January 1, 2006. Since this drug coverage will be available, Prescription Advantage will provide supplemental coverage for prescription drug purchases of members who are eligible for Medicare. For all members, with or without Medicare, Prescription Advantage will only cover classes of drugs that Medicare has not excluded from coverage. We have determined that you have taken one or more of the prescription drugs listed on the second page of this letter, which Medicare has excluded from coverage. As of March 31, 2006, Prescription Advantage will no longer cover these drugs. We recommend that you speak to your doctor regarding this change in drug coverage as he or she can most appropriately advise you of any viable alternatives or substitutions that are available to you. Your doctor is the most qualified person to determine which prescription drugs are suitable for you. If you have any questions, please call Customer Service (toll free) at 1-800-AGE-INFO (1-800- 243-4636) or TTY for the deaf and hard of hearing at 1-877-610-0241. Sincerely, Prescription Advantage Commonwealth of Massachusetts Executive Office of Health and Human Services Executive Office of Elder Affairs Prescription Advantage 1-800-AGE-INFO (1-800-243-4636) TTY: 1-877-610-0241 www.800ageinfo.com 3/9/2006 Page 2 of 3 Alkabel-sr tablet Fioricet tablet Phenazopyridine plus -

Safety Data Sheet
SDS Number 110529 Approved/Revised 23-Jun-2008 Version 14 Material AGENERASE SOFTGEL CAPSULES SAFETY DATA SHEET 1. IDENTIFICATION OF THE SUBSTANCE/PREPARATION AND OF THE COMPANY/UNDERTAKING space Material AGENERASE SOFTGEL CAPSULES space Synonym(s) AGENERASE CAPSULES * AGENERASE SOFTGEL CAPSULES 50 MG * AGENERASE SOFTGEL CAPSULES 150 MG * AMPRENAVIR, FORMULATED PRODUCT space Company Name GlaxoSmithKline, Corporate Environment, Health & Safety 980 Great West Road Brentford, Middlesex TW8 9GS UK space UK General Information: +44-20-8047-5000 Transport Emergency (EU) +44-1865-407333 Medical Emergency +1-612-221-3999, Ext 221 Information and Advice: US number, available 24 hours Multi-language response space GlaxoSmithKline, Corporate Environment, Health & Safety One Franklin Plaza, 200 N 16th Street Philadelphia, PA 19102-1225 US space US General Information: +1-888-825-5249 Transport Emergency (non EU) +1-703-527-3887 US number, available 24 hours Multi-language response . 2. COMPOSITION / INFORMATION ON INGREDIENTS space Ingredients CAS # Percent EC-No. AMPRENAVIR 161814-49-9 6 to 7.4 NON-HAZARDOUS INGREDIENTS Unassigned 92.6 to 94 3. HAZARDS IDENTIFICATION space Fire and Explosion Expected to be non-combustible. space Health Caution - Pharmaceutical agent. May produce adverse effects on the development of human offspring. Possible effects of overexposure in the workplace include: diarrhoea; nausea; tingling; vomiting; rash. Health effects information is based on hazards of components. Handling this product in its final form presents minimal risk from occupational exposure. space Environment No information is available about the potential of this product to produce adverse environmental effects. 4. FIRST-AID MEASURES space Ingestion Never attempt to induce vomiting. -

From Molecule to Dose Form Bioavailability Toolkit to Fast Track Development Introduction
contents introduction 2 part one: Connecting the 4 Dots in Drug Delivery part two: The Bioavailability 8 Toolbox part three: The Toolbox of 2025 16 part four: Delivering Change 21 from molecule to dose form bioavailability toolkit to fast track development introduction When it comes to drug formulation challenges, there trend and the key to succeed in this competitive is no one-size-fits-all solution. landscape. The leading evidence of this trend includes more direct-to-consumer advertising and a The 3rd annual drug delivery landscape survey* recent patient focused drug development initiative confirms that bioavailability, safety, efficacy by the FDA to gather the patient’s perspective and solubility are the top challenges faced by on their condition and available treatments. formulation scientists. The pharmaceutical and Interestingly, survey data shows that although biopharmaceutical industries have to overcome not the industry has identified the value of patient- only formulation challenges but also the business centric drug design, it has not been very successful challenges of bringing a product successfully to in applying it. For instance, to successfully apply market. The survey participants, a vast majority of patient-centric design, the drug product should be whom are Formulation Scientists and R&D Managers, adapted as per the patient’s preference at earlier indicated that development time, selecting the stages of drug development to avoid high costs at appropriate drug delivery platform, budget and late stages, a practice which is not very common in choosing the right external partner are the top the industry. Also, when asked to rate the significant business challenges. -

Novel Thyroxine Formulations: a Further Step Toward Precision Medicine
Endocrine (2019) 66:87–94 https://doi.org/10.1007/s12020-019-02049-x REVIEW Novel thyroxine formulations: a further step toward precision medicine 1 2 1,3 Camilla Virili ● Pierpaolo Trimboli ● Marco Centanni Received: 3 June 2019 / Accepted: 5 August 2019 © Springer Science+Business Media, LLC, part of Springer Nature 2019 Abstract Levothyroxine (T4) is a critical-dose drug, because little variations in the blood concentration may cause treatment failure as well as iatrogenic thyrotoxicosis. Despite the dose response of this drug being more carefully titrated nowadays, several papers still report that a significant fraction of patients treated with levothyroxine demonstrate a TSH which is not on target. Moreover, some widespread gastrointestinal disorders as well as interfering drugs and foods may cause the “refractoriness” of a significant number of patients to an expected dose of thyroxine. The increasing awareness of the mechanisms interfering with the oral thyroid hormone bioavailability and the body of evidence regarding the complexity of treatment in certain classes of patients prompted pharmaceutical research to identify new hormonal formulations to optimize the performance of fi 1234567890();,: 1234567890();,: this drug. In this brief review, the progression of the scienti c knowledge of novel T4 formulations use has been analyzed. Keywords Liquid levothyroxine ● Softgel thyroxine ● Thyroxine absorption ● Thyroxine malabsorption ● Drugs dissolution ● Gastrointestinal disorders Introduction levothyroxine doses are used to treat patients of different ages and with specific clinical conditions [8]. Synthesized in 1927 by Harrington [1] and obtained as The daily requirement of levothyroxine depends on sodium salt in 1950 [2], levothyroxine (T4) has subse- patients’ lean body mass [9], the leading cause of hypo- quently obtained the role of gold standard in the treatment thyroidism and the therapeutic goal [e.g. -

Important Pharmacy Information There Is No Copay When Your Primary Care Provider (PCP) Or Unitedhealthcare Community Plan Specialist Writes You a Covered Prescription
Important Pharmacy Information There is no copay when your Primary Care Provider (PCP) or UnitedHealthcare Community Plan Specialist writes you a covered prescription. But you can get many over-the-counter (OTC) medicines free when you have a prescription. You can get the medications listed on the following pages when they are medically necessary and you get a written prescription from your UnitedHealthcare Community Plan doctor and take it to a UnitedHealthcare Community Plan pharmacy. To get your medicine: • Take your prescription to a UnitedHealthcare Community Plan pharmacy. To find a pharmacy, call 1-800-903-5253 or go to UHCCommunityPlan.com. • For your safety, we urge you to select a single pharmacy from which to get your drugs. • Get to know the pharmacist and build a relationship. If the UnitedHealthcare Community Plan pharmacy says they cannot fill your covered prescription or you have to pay more than your copay: Do not leave the pharmacy. Do not pay for it yourself. Ask the pharmacy why they cannot fill your prescription. Response Your Solution Not Covered • Ask them to call OptumRx right away to find out which medicine is covered. • Ask them to call your doctor to see if you can get the covered medicine instead. Prior • Ask them to call your doctor for a prior authorization. Authorization • You can call your doctor and ask that a prior authorization be sent to: Needed UnitedHealthcare Pharmacy Prior Notification Service Fax 1-866-940-7328 Phone 1-800-310-6826 Refill Too Soon • Ask what day it can be filled. • Pick your prescription up the day it can be filled. -

SOFT GELATIN CAPSULES Capsules Are Solid Oral Dosage Forms in Which the Drug Is Enclosed Within a Hard Or Soft Shell
CAPSULES PRODUCTION. MICROCAPSULES. HARD AND SOFT GELATIN CAPSULES Capsules are solid oral dosage forms in which the drug is enclosed within a hard or soft shell. The shell is normally made from gelatin and results in a simple, easy - to - swallow formulation with no requirement for a further coating step. They can be either hard or soft depending on the nature of the capsule shell, with soft capsules possessing a fl exible, plasticized gelatin fi lm. Hard gelatin capsules are usually rigid two - piece capsules that are manufactured in one procedure and packed in another totally separate operation, whereas the formulation of soft gelatin capsules is more complex but all steps are integrated. There is a growing interest in using non - animal - derived products for formulation of the capsule shells to address cultural, religious, and dietary requirements. HPMC (e.g., V - caps, Quali - VC, Vegicaps) and pullulan shells (NPCaps) and starch are alternatives. Hard - Shell Gelatin Capsules Although the challenges of powder blending, homogeneity, and lubcricity exist for capsules as for tablets, they are generally perceived to be a more fl exible formulation as there is no requirement for the powders to form a robust compact. This means that they may also be more suitable for delivery of granular and beadlike formulations, fragile formulations that could be crushed by the normal compaction step. They are commonly employed in clinical trials due to the relative ease of blinding and are useful for taste masking. Capsules are usually more expensive dosage forms than an equivalent tablet formulation due to the increased cost of the shells and the slower production rates. -

Challenges of Dissolution Methods Development for Soft Gelatin Capsules
pharmaceutics Review Challenges of Dissolution Methods Development for Soft Gelatin Capsules Festo Damian 1, Mohammad Harati 2, Jeff Schwartzenhauer 3, Owen Van Cauwenberghe 1,4 and Shawn D. Wettig 1,5,* 1 School of Pharmacy, University of Waterloo, 200 University Ave W, Waterloo, ON N2L 3G1, Canada; [email protected] (F.D.); [email protected] (O.V.C.) 2 Department of Natural Sciences, University of Michigan, 2209 Natural Sciences Building, 4901 Evergreen Rd, Dearborn, MI 48128, USA; [email protected] 3 Catalent Pharma Solutions, 2125 Ambassador Dr, Windsor, ON N9C 3R5, Canada; [email protected] 4 Bio Therapeutic Molecules Inc., 70 Southgate Drive, Unit 4, Guelph, ON N1G 4P5, Canada 5 Waterloo Institute for Nanotechnology, University of Waterloo, 200 University Ave W, Waterloo, ON N2L 3G1, Canada * Correspondence: [email protected]; Tel.: +1-519-888-4567 (ext. 42221) Abstract: Recently, the development of soft gelatin capsules (SGCs) dosage forms has attracted a great deal of interest in the oral delivery of poorly water-soluble drugs. This is attributed to the increased number of poorly soluble drugs in the pipeline, and hence the challenges of finding innovative ways of developing bioavailable and stable dosage forms. Encapsulation of these drugs into SGCs is one of the approaches that is utilized to deliver the active ingredients to the systemic circulation to overcome certain formulation hurdles. Once formulated, encapsulated drugs in the form of SGCs require suitable in vitro dissolution test methods to ensure drug product quality and Citation: Damian, F.; Harati, M.; Schwartzenhauer, J.; Van performance. This review focuses on challenges facing dissolution test method development for Cauwenberghe, O.; Wettig, S.D. -

Applications of Polymer Blends in Drug Delivery Nasser N
Nyamweya Future Journal of Pharmaceutical Sciences (2021) 7:18 Future Journal of https://doi.org/10.1186/s43094-020-00167-2 Pharmaceutical Sciences REVIEW Open Access Applications of polymer blends in drug delivery Nasser N. Nyamweya Abstract Background: Polymers are essential components of many drug delivery systems and biomedical products. Despite the utility of many currently available polymers, there exists a demand for materials with improved characteristics and functionality. Due to the extensive safety testing required for new excipient approval, the introduction and use of new polymers is considerably limited. The blending of currently approved polymers provides a valuable solution by which the limitations of individual polymers can be addressed. Main body: Polymer blends combine two or more polymers resulting in improved, augmented, or customized properties and functionality which can result in significant advantages in drug delivery applications. This review discusses the rationale for the use of polymer blends and blend polymer-polymer interactions. It provides examples of their use in commercially marketed products and drug delivery systems. Examples of polymer blends in amorphous solid dispersions and biodegradable systems are also discussed. A classification scheme for polymer blends based on the level of material processing and interaction is presented. Conclusion: The use of polymer blends represents a valuable and under-utilized resource in addressing a diverse range of drug delivery challenges. It is anticipated that new drug molecule development challenges such as bioavailability enhancement and the demand for enabling excipients will lead to increased applications of polymer blends in pharmaceutical products. Keywords: Polymer blends, Drug delivery, Dosage forms, Solid dispersions, Bioavailability enhancement Background that the amounts used are within the limits for which Polymers are widely used in the formulation of pharma- safety has been established. -
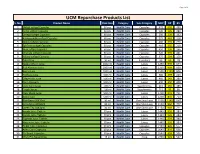
UCM Repurchase Products List S
Page 1 of 4 UCM Repurchase Products List S. No. Product Name Pack Size Category Sub-Category MRP DP BV 1 Shilajit softgel Capsules 60 pcs Health Care Capsules 975 699 490 2 Garlic softgel Capsules 60 pcs Health Care Capsules 500 399 280 3 Omega softgel Capsules 60 pcs Health Care Capsules 380 299 192 4 Ashwagandha softgel Capsules 60 pcs Health Care Capsules 525 449 318 5 Neem softgel Capsules 60 pcs Health Care Capsules 480 399 279 6 Spiriluna softgel Capsules 60 pcs Health Care Capsules 410 350 210 7 Noni softgel Capsules 60 pcs Health Care Capsules 450 399 266 8 Moringa softgel Capsules 60 pcs Health Care Capsules 720 599 441 9 Kalonji softgel Capsules 60 pcs Health Care Capsules 375 299 150 10 Tulsi Drop 30 ml Health Care Essentials 90 60 28 11 Seabuckthorn Juice 1000 ml Health Care Juices 1250 999 644 12 Red Aloevera Juice 1000 ml Health Care Juices 1250 999 600 13 Berry Juice 1000 ml Health Care Juices 1500 1199 806 14 Triphala Juice 500 ml Health Care Juices 380 299 223 15 Giloy+Tulsi Juice 500 ml Health Care Juices 460 349 280 16 Four Aloevera 500 ml Health Care Juices 600 450 408 17 Liver Care Syrup 100 ml Health Care Suppliments 215 185 185 18 Cough Syrup 100 ml Health Care Suppliments 115 95 80 19 Clear Block Juice 500 ml Health Care Juices 1400 949 711 20 Gynic Care 500 ml Health Care Juices 1200 850 753 21 Blue Berry 30X30ml 30 ml Health Care One Shot Juice 1500 1249 1007 22 Amla Mint 30X30ml 30 ml Health Care One Shot Juice 1150 960 777 23 Green Tea 30X30ml 30 ml Health Care One Shot Juice 1150 949 711 24 Amala Juice Tablets 30 pcs Health Care Juices 1440 599 384 25 Karela Juice Tablets 30 pcs Health Care Juices 1440 599 384 26 Triphala Juice Tablets 30 pcs Health Care Juices 1440 599 384 27 Alovevera Juice Tablets 30 pcs Health Care Juices 1440 599 384 28 Sugar Care Capsules 60 pcs Health Care Capsules 2160 899 577 29 Artho Care Capsules 60 pcs Health Care Capsules 2640 899 506 30 Re-Boost Capsules 30 pcs Health Care Capsules 2640 899 506 31 Wom Pro Nasal Drop 15 ml Health Care Drop 2700 949 547 Page 2 of 4 S. -

US5573756.Pdf
|||||||| US005573756A United States Patent (19) 11 Patent Number: 5,573,756 Lambrechts (45) Date of Patent: Nov. 12, 1996 (54) SHAMPOO CONDITIONER AND SOFTGEL 5,089,269 2/1992 Noda et al. ............................. 424/456 FILLED THEREWITH OTHER PUBLICATIONS 75 Inventor: John Lambrechts, Burbank, Calif. "Cocamide DEA,”International Cosmetic Ingredient Dictio (73) Assignee: paper Pharmacaps Inc., Chatsworth, "PEG-Glycerylnary, Fifth Edition, Cocoate"International Washington, D.C. (1993), Cosmetic vol. Ingredient 1, p. 157. 1. Dictionary, Fifth Edition, Washington, D.C. (1993), vol. 1, p. 491. (21) Appl. No. 377,378 "Polyguaternium-10.” International Cosmetic Ingredient (22 Filed: Jan. 25, 1995 Dictionary, Fifth Edition, Washington, D.C. (1993), vol. 1, p. 571. (51) Int. Cl. ................... A61K 7/075; A61K9/48 52 U.S. Cl. .................. 424/70.24; 424/70.1; 424/70.22; Primary Examiner-Jyothsna Venkat 424/456 Attorney, Agent, or Firm-Jeffer, Mangels, Butler & Mar (58) Field of Search ..................................... 424/456, 401, maro LLP 424/70.1, 70.22, 70.24 (57) ABSTRACT (56) References Cited An shampoo conditioner composition which is capable of U.S. PATENT DOCUMENTS being stably encapsulated within a gelatin shell includes a concentrated surfactant, a cationic conditioner, and a carrier, 3,590,122 6/1971 Hutcheson et al. ....................... 424/70 and has an equilibrium relative humidity (ERH) of less than 3,808,329 4/1974 Bolich, Jr. et al. .... ... 424/70 about 65%. 3,816,616 6/1974 Anguillo et al. ....... ... 424/70 4,272,515 6/1981 Hofman et al..... ... 424/70 4,701,322 10/1987 Dixon et al. .............................. 424/70 19 Claims, No Drawings 5,573,756 1 2 SHAMPOO CONDITIONER AND SOFTGEL with water, working the composition through the hair, and FLLED THEREWITH rinsing the composition from the hair. -

Patient Acceptability and Satisfaction with a Low-Dose Solubilized Vaginal Estradiol Softgel Capsule, TX-004HR
CE: A.M.; MENO-D-16-00342; Total nos of Pages: 6; MENO-D-16-00342 Menopause: The Journal of The North American Menopause Society Vol. 24, No. 8, pp. 000-000 DOI: 10.1097/GME.0000000000000848 ß 2017 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of The North American Menopause Society. Patient acceptability and satisfaction with a low-dose solubilized vaginal estradiol softgel capsule, TX-004HR Sheryl A. Kingsberg, PhD,1 Robin Kroll, MD,2 Irwin Goldstein, MD,3 Harvey Kushner, PhD,4 Brian Bernick, MD,5 Shelli Graham, PhD,5 Sebastian Mirkin, MD,5 and Ginger D. Constantine, MD6 Abstract Objective: TX-004HR is an investigational, muco-adhesive, vaginal, softgel capsule containing low-dose, solubilized, 17b-estradiol designed to treat postmenopausal vulvar and vaginal atrophy (VVA) and improve user experience without an applicator and less mess. Methods: As part of the 12-week, placebo-controlled, double-blind, phase 3 REJOICE trial evaluating the efficacy/safety of 4-, 10-, and 25-mg TX-004HR in 764 postmenopausal women with VVA, a five-question product survey was administered. Pearson correlation coefficients were used to evaluate correlations between clinical endpoints (vaginal physiology, dyspareunia, and vaginal dryness) and patient acceptability and satisfaction. Results: Majority of the women receiving TX-004HR or placebo reported that the product was easy to use (85.4%-92.1%) and rated ease of capsule insertion as ‘‘good’’ to ‘‘excellent’’ (75.0%-82.6%). A significantly greater percentage of women reported being ‘‘very satisfied’’ or ‘‘satisfied’’ with TX-004HR (68.6%-76.3%) than with placebo (56.8%, P < 0.05 for all).