Molecular Identification of Roots from a Grassland Community Using Size
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Vascular Plants of Massachusetts
The Vascular Plants of Massachusetts: The Vascular Plants of Massachusetts: A County Checklist • First Revision Melissa Dow Cullina, Bryan Connolly, Bruce Sorrie and Paul Somers Somers Bruce Sorrie and Paul Connolly, Bryan Cullina, Melissa Dow Revision • First A County Checklist Plants of Massachusetts: Vascular The A County Checklist First Revision Melissa Dow Cullina, Bryan Connolly, Bruce Sorrie and Paul Somers Massachusetts Natural Heritage & Endangered Species Program Massachusetts Division of Fisheries and Wildlife Natural Heritage & Endangered Species Program The Natural Heritage & Endangered Species Program (NHESP), part of the Massachusetts Division of Fisheries and Wildlife, is one of the programs forming the Natural Heritage network. NHESP is responsible for the conservation and protection of hundreds of species that are not hunted, fished, trapped, or commercially harvested in the state. The Program's highest priority is protecting the 176 species of vertebrate and invertebrate animals and 259 species of native plants that are officially listed as Endangered, Threatened or of Special Concern in Massachusetts. Endangered species conservation in Massachusetts depends on you! A major source of funding for the protection of rare and endangered species comes from voluntary donations on state income tax forms. Contributions go to the Natural Heritage & Endangered Species Fund, which provides a portion of the operating budget for the Natural Heritage & Endangered Species Program. NHESP protects rare species through biological inventory, -

45Th Anniversary Year
VOLUME 45, NO. 1 Spring 2021 Journal of the Douglasia WASHINGTON NATIVE PLANT SOCIETY th To promote the appreciation and 45 conservation of Washington’s native plants Anniversary and their habitats through study, education, Year and advocacy. Spring 2021 • DOUGLASIA Douglasia VOLUME 45, NO. 1 SPRING 2021 journal of the washington native plant society WNPS Arthur R. Kruckberg Fellows* Clay Antieau Lou Messmer** President’s Message: William Barker** Joe Miller** Nelsa Buckingham** Margaret Miller** The View from Here Pamela Camp Mae Morey** Tom Corrigan** Brian O. Mulligan** by Keyna Bugner Melinda Denton** Ruth Peck Ownbey** Lee Ellis Sarah Reichard** Dear WNPS Members, Betty Jo Fitzgerald** Jim Riley** Mary Fries** Gary Smith For those that don’t Amy Jean Gilmartin** Ron Taylor** know me I would like Al Hanners** Richard Tinsley Lynn Hendrix** Ann Weinmann to introduce myself. I Karen Hinman** Fred Weinmann grew up in a small town Marie Hitchman * The WNPS Arthur R. Kruckeberg Fellow Catherine Hovanic in eastern Kansas where is the highest honor given to a member most of my time was Art Kermoade** by our society. This title is given to Don Knoke** those who have made outstanding spent outside explor- Terri Knoke** contributions to the understanding and/ ing tall grass prairie and Arthur R. Kruckeberg** or preservation of Washington’s flora, or woodlands. While I Mike Marsh to the success of WNPS. Joy Mastrogiuseppe ** Deceased love the Midwest, I was ready to venture west Douglasia Staff WNPS Staff for college. I earned Business Manager a Bachelor of Science Acting Editor Walter Fertig Denise Mahnke degree in Wildlife Biol- [email protected] 206-527-3319 [email protected] ogy from Colorado State Layout Editor University, where I really Mark Turner Office and Volunteer Coordinator [email protected] Elizabeth Gage got interested in native [email protected] plants. -

Propagation of Colorado Natives at Little Valley©
224 Combined Proceedings International Plant Propagators’ Society, Volume 58, 2008 Propagation of Colorado Natives at Little Valley © Brian Core Little Valley Wholesale Nursery, 13022 E. 136th Ave., Brighton, Colorado 80601 U.S.A. Email: [email protected] INTRODUCTION Little Valley Wholesale Nursery was established in 1979 with 15 acres of field and container stock and a staff of four people. Over the years, it has expanded to en- compass 141 acres and a peak-season staff of more than 150 people. The plant palette now includes more than 500 taxa of perennials, 275 taxa of shrubs, and over 130 taxa of trees. The area served by Little Valley’s distribution system includes Wyoming, Colorado, New Mexico, and Arizona. Little Valley’s motto “The Rocky Mountain Standard” expresses the commitment to be the best wholesale plant dis- tributor in the western region. Native plants have been an important part of Little Valley’s plant mix for many years. Native plants are ideal for low-input sustainable landscapes. The Denver Metro area receives 8–15 inches of rain per year; in contrast, many eastern cities receive 50 or even 60 inches of annual precipitation. Colorado has a rapidly expanding population that threatens to deplete the water supply within a few decades. Many native plants require no supplemental water when established in the landscape; water that is not wasted on bluegrass lawns is water that can be used for human consumption. Native plants also require less frequent fertilizer and pesticide appli- cations; overuse of fertilizer and pesticide can lead to contamination of groundwater supplies with carcinogens. -

Legumes of the North-Central States: C
LEGUMES OF THE NORTH-CENTRAL STATES: C-ALEGEAE by Stanley Larson Welsh A Dissertation Submitted, to the Graduate Faculty in Partial Fulfillment of The Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Subject: Systematic Botany Approved: Signature was redacted for privacy. Signature was redacted for privacy. artment Signature was redacted for privacy. Dean of Graduat College Iowa State University Of Science and Technology Ames, Iowa I960 ii TABLE OF CONTENTS Page ACKNOWLEDGMENTS iii INTRODUCTION 1 HISTORICAL ACCOUNT 3 MATERIALS AND METHODS 8 TAXONOMIC AND NOMENCLATURE TREATMENT 13 REFERENCES 158 APPENDIX A 176 APPENDIX B 202 iii ACKNOWLEDGMENTS The writer wishes to express his deep gratitude to Professor Duane Isely for assistance in the selection of the problem and for the con structive criticisms and words of encouragement offered throughout the course of this investigation. Support through the Iowa Agricultural Experiment Station and through the Industrial Science Research Institute made possible the field work required in this problem. Thanks are due to the curators of the many herbaria consulted during this investigation. Special thanks are due the curators of the Missouri Botanical Garden, U. S. National Museum, University of Minnesota, North Dakota Agricultural College, University of South Dakota, University of Nebraska, and University of Michigan. The cooperation of the librarians at Iowa State University is deeply appreciated. Special thanks are due Dr. G. B. Van Schaack of the Missouri Botanical Garden library. His enthusiastic assistance in finding rare botanical volumes has proved invaluable in the preparation of this paper. To the writer's wife, Stella, deepest appreciation is expressed. Her untiring devotion, work, and cooperation have made this work possible. -

Terrestrial Ecological Classifications
INTERNATIONAL ECOLOGICAL CLASSIFICATION STANDARD: TERRESTRIAL ECOLOGICAL CLASSIFICATIONS Alliances and Groups of the Central Basin and Range Ecoregion Appendix Accompanying Report and Field Keys for the Central Basin and Range Ecoregion: NatureServe_2017_NVC Field Keys and Report_Nov_2017_CBR.pdf 1 December 2017 by NatureServe 600 North Fairfax Drive, 7th Floor Arlington, VA 22203 1680 38th St., Suite 120 Boulder, CO 80301 This subset of the International Ecological Classification Standard covers vegetation alliances and groups of the Central Basin and Range Ecoregion. This classification has been developed in consultation with many individuals and agencies and incorporates information from a variety of publications and other classifications. Comments and suggestions regarding the contents of this subset should be directed to Mary J. Russo, Central Ecology Data Manager, NC <[email protected]> and Marion Reid, Senior Regional Ecologist, Boulder, CO <[email protected]>. Copyright © 2017 NatureServe, 4600 North Fairfax Drive, 7th floor Arlington, VA 22203, U.S.A. All Rights Reserved. Citations: The following citation should be used in any published materials which reference ecological system and/or International Vegetation Classification (IVC hierarchy) and association data: NatureServe. 2017. International Ecological Classification Standard: Terrestrial Ecological Classifications. NatureServe Central Databases. Arlington, VA. U.S.A. Data current as of 1 December 2017. Restrictions on Use: Permission to use, copy and distribute these data is hereby granted under the following conditions: 1. The above copyright notice must appear in all documents and reports; 2. Any use must be for informational purposes only and in no instance for commercial purposes; 3. Some data may be altered in format for analytical purposes, however the data should still be referenced using the citation above. -

Vascular Plants of Kluane
26 Blueleaved strawberry Fragaria virginiana 63 Greyleaf willow Salix glauca Kluane National Park and Reserve 27 Bog blueberrry Vaccinium uliginosum 64 Ground cedar, Lycopodium complanatum 28 Bog labrador-tea Ledum groenlandica Creeping jenny 65 Hairy rockcress Arabis hirsuta 29 Boreal aster Aster alpinus 30 Boreal wormwood Artemisia arctica 66 Heart-leaf listera Listera borealis Vascular 31 Bristly stickseed Lappula myosotis 67 Heartleaf arnica Arnica cordifolia 32 Broadglumed wheatgrass Agropyron trachycaulum 68 High bush cranbery Viburnum edule Plants List 33 Broadleaf lupine Lupinus arcticus 69 Holboell's rockcress Arabis holboellii 34 Buffaloberry, Soapberry Sheperdia canadensis 70 Horned dandelion Taraxacum lacerum 35 Canada butterweed Senecio pauperculus 71 Kotzebue's grass-of- Parnassia kotzebuei 36 Chestnut rush Juncus castaneus parnassus 1 Alaska moss heath Cassiope stelleriana 37 Cleft-leaf groundsel Senecio streptanthifolius 72 Kuchei's lupine Lupinus kuschei 2 Alaska willow Salix alaxensis 38 Common horsetail Equisetum arvense 73 Labrador lousewort Pedicularis labradorica 3 Alkali bluegrass Poa juncifolia 39 Common mountain Juniperus communis 74 Lance-leaved draba Draba lanceolata 4 Alkali grass Puccinellia interior juniper 75 Lanceleaved stonecrop Sedum lanceolatum 5 Alpine bluegrass Poa alpina 40 Cow parsnip Heracleum lanatum 76 Lapland cassiope Cassiope tetragona 6 Alpine fescue Festuca ovina 41 Creeping juniper Juniperus horizontalis 77 Leafless pyrola Pyrola asarifolia 7 Alpine milk-vetch Astragalus alpinus 42 Creeping -

Easton Collection Center Living Roof Families: 31 Genera: 83 Species: 102 Total Taxa: 104
Easton Collection Center Living Roof Families: 31 Genera: 83 Species: 102 Total Taxa: 104 Achillea Millefolium Achnatherum hymenoidea Aegilops cylindrica Common yarrow Indian Rice Grass Jointed Goat Grass Allium cernuum Amaranthus albus Amaranthus powellii Nodding Onion Tumbleweed Green Amaranth Ambrosia tomentosa Antennaria parvifolia Arenaria fendleri Skeleton-Leaf Burr-Ragweed Little-Leaf pussytoes Fendler’s Matted Sandwort Artemisia dracunculus Artemisia ludoviciana Asclepias subverticillata Dragon Wormwood White Sagebrush Horsetail Milkweed Astragalus humistratus Bahia dissecta Bidens tenuisecta Ground-cover milk-vetch Sunray daisy Slim-Love Beggarticks Blepharoneuron tricholepis Bouteloua curtipendula Bouteloua gracilis Pine-Dropseed Side-Oats Grama Blue Grama 2 Bouteloua simplex Bromus tectorum Capsella bursa-pastoris Matted Grama Cheat grass Shepherd’s-Purse Castillega integra Chamaesyce serpyllifolia Chenopodium leptophyllum Squawfeather Thyme-Leaf Sandmat Narrow-Leaf Goosefoot Cirsium arizonicum Cleome serrulata Conyza canadensis Arizona Thistle Rocky Mountain Beeplant Canadian Horseweed 3 Echinocereus fendleri Elymus elymoides Epilobium brachycarpum Pink-Flower Hedgehog Cactus Western Bottle-Brush Grass Tall Annual Willowherb Eragrostis pectinacea Erigeron divergens Erigeron flagellaris Purple Love Grass Spreading Fleabane Trailing Fleabane Erodium cicutarium Fallugia paradoxa Festuca arizonica Red-Stem Stork’s-Bill Apache-Plume Arizona Fescue 4 Festuca ovina Gaillardia aristata Gaura coccinea Sheep Fescue Great Blanket-Flower -
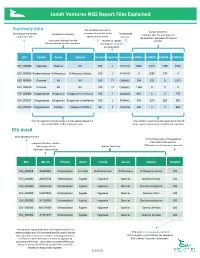
NGS Report Explained
Jonah Ventures NGS Report Files Explained Summary data The % of base pairs in the Sample identifiers Exact Sequence Variant sequence that match to the The detected Exact Sequence Variant Consensus Taxonomy The detected Numbers after the decimal point unique identifier species in the library. sequence. represent lab replicates of the same Taxonomic ranking from the Number of species sample. library matched to each sequence. matching the sequence at a given level. ESV Family Genus Species %match # Species Sequence S10001.1 S10002.1 S10003.1 S10003.2 ESV_000080 Fagaceae Quercus NA 100 7 AAAAAG… 3066 1340 5780 3462 ESV_000818 Orobanchaceae Orthocarpus Orthocarpus luteus 100 1 AAAAAG… 0 2582 249 0 ESV_000006 Poaceae NA NA 100 77 GAAAAG… 584 101 0 1039 ESV_000050 Poaceae NA NA 100 12 GAAAAG… 1328 0 0 0 ESV_000308 Polygonaceae Polygonum Polygonum humifusum 100 2 AAAAAG… 902 0 0 278 ESV_000027 Polygonaceae Eriogonum Eriogonum umbellatum 100 1 ATAAAG… 341 124 282 381 ESV_000520 Polygonaceae Fallopia Fallopia multiflora 99 1 AAAAAG… 126 0 0 994 “NA” at a taxonomic ranks means multiple species present in The number in each cell is the absolute number of the sample differ in that taxonomic rank times a given sequence was read by the sequencer. ESV detail Exact Sequence Variant The % of base pairs in the sequence that match to the species. Index identification number. that match to the species. Each sample has an Species Taxonomy 100% indicates all base pairs matched individual index number. ESV GB_ID Phylum Order Family Genus Species %match ESV_000818 -

An Ethnobotany of Mount Rushmore National Memorial
University of Massachusetts Amherst ScholarWorks@UMass Amherst Landscape Architecture & Regional Planning Masters Projects Landscape Architecture & Regional Planning Summer 2019 An Ethnobotany of Mount Rushmore National Memorial Meredith Savage University of Massachusetts Amherst Follow this and additional works at: https://scholarworks.umass.edu/larp_ms_projects Part of the Landscape Architecture Commons Savage, Meredith, "An Ethnobotany of Mount Rushmore National Memorial" (2019). Landscape Architecture & Regional Planning Masters Projects. 92. Retrieved from https://scholarworks.umass.edu/larp_ms_projects/92 This Article is brought to you for free and open access by the Landscape Architecture & Regional Planning at ScholarWorks@UMass Amherst. It has been accepted for inclusion in Landscape Architecture & Regional Planning Masters Projects by an authorized administrator of ScholarWorks@UMass Amherst. For more information, please contact [email protected]. AN ETHNOBOTANY OF MOUNT RUSHMORE NATIONAL MEMORIAL Department of Landscape Architecture and Regional Planning University of Massachusetts Amherst Master of Regional Planning Project Meredith S. Savage July 2019 © Copyright by Meredith S. Savage 2019 All Rights Reserved AN ETHNOBOTANY OF MOUNT RUSHMORE NATIONAL MEMORIAL Department of Landscape Architecture and Regional Planning University of Massachusetts Amherst Master of Regional Planning Project Meredith S. Savage July 2019 Approved as to style and content by: __________________________________________ Dr. Elizabeth Brabec, -

Survey and Analysis of Plant Community Types of Writing-On-Stone Provincial Park
Survey and Analysis of Plant Community Types of Writing-on-Stone Provincial Park Survey and Analysis of Plant Community Types of Writing-on-Stone Provincial Park FINAL REPORT Prepared for Alberta Sustainable Resource Development Resource Data Branch Edmonton, Alberta Prepared by Wildlands Ecological Consulting Ltd. #60 Neal Close, Red Deer, AB T4P 1N4 Office: (403) 346-1057 Fax: (403) 346-3257 March 29, 2004 ABSTRACT Identification and monitoring of biological diversity in Alberta is the primary mandate of Alberta Natural Heritage Information Centre (ANHIC). A fundamental strategy in the preservation of functional ecosystems is to identify and preserve a full compliment of habitat types, which in return support a diversity of animals, plants, and other life forms. Vegetation is an integral component of habitat and provides a relatively easy means of inventory and monitoring of ecosystem health both spatially and temporally. The current study, in Writing-on-Stone Provincial Park, focuses on the identification of unique riparian and coulee plant associations, or plant community type. The study area is situated approximately 8 km north of the USA border and 40 km east of the town of Milk River and occurs at the interface between the Mixedgrass and Dry Mixedgrass Natural Subregions. Priority research is currently required to address gaps in the identification, description and distribution of plant associations within the coulee and riparian zones of these Subregions. Many of these communities recur over the landscape but may occur as very small patches of 1m2 or less. Alberta Sustainable Resource Development (ASRD) Resource Data Division contracted Wildlands Ecological to complete an inventory of natural/semi-natural plant communities, statistically analyse the data, and describe potential plant community or association types. -

Vascular Plants and a Brief History of the Kiowa and Rita Blanca National Grasslands
United States Department of Agriculture Vascular Plants and a Brief Forest Service Rocky Mountain History of the Kiowa and Rita Research Station General Technical Report Blanca National Grasslands RMRS-GTR-233 December 2009 Donald L. Hazlett, Michael H. Schiebout, and Paulette L. Ford Hazlett, Donald L.; Schiebout, Michael H.; and Ford, Paulette L. 2009. Vascular plants and a brief history of the Kiowa and Rita Blanca National Grasslands. Gen. Tech. Rep. RMRS- GTR-233. Fort Collins, CO: U.S. Department of Agriculture, Forest Service, Rocky Mountain Research Station. 44 p. Abstract Administered by the USDA Forest Service, the Kiowa and Rita Blanca National Grasslands occupy 230,000 acres of public land extending from northeastern New Mexico into the panhandles of Oklahoma and Texas. A mosaic of topographic features including canyons, plateaus, rolling grasslands and outcrops supports a diverse flora. Eight hundred twenty six (826) species of vascular plant species representing 81 plant families are known to occur on or near these public lands. This report includes a history of the area; ethnobotanical information; an introductory overview of the area including its climate, geology, vegetation, habitats, fauna, and ecological history; and a plant survey and information about the rare, poisonous, and exotic species from the area. A vascular plant checklist of 816 vascular plant taxa in the appendix includes scientific and common names, habitat types, and general distribution data for each species. This list is based on extensive plant collections and available herbarium collections. Authors Donald L. Hazlett is an ethnobotanist, Director of New World Plants and People consulting, and a research associate at the Denver Botanic Gardens, Denver, CO. -

Riverside State Park
Provisonal Report Rare Plant and Vegetation Survey of Riverside State Park Pacific Biodiversity Institute 2 Provisonal Report Rare Plant and Vegetation Survey of Riverside State Park Peter H. Morrison [email protected] George Wooten [email protected] Juliet Rhodes [email protected] Robin O’Quinn, Ph.D. [email protected] Hans M. Smith IV [email protected] January 2009 Pacific Biodiversity Institute P.O. Box 298 Winthrop, Washington 98862 509-996-2490 Recommended Citation Morrison, P.H., G. Wooten, J. Rhodes, R. O’Quinn and H.M. Smith IV, 2008. Provisional Report: Rare Plant and Vegetation Survey of Riverside State Park. Pacific Biodiversity Institute, Winthrop, Washington. 433 p. Acknowledgements Diana Hackenburg and Alexis Monetta assisted with entering and checking the data we collected into databases. The photographs in this report were taken by Peter Morrison, Robin O’Quinn, Geroge Wooten, and Diana Hackenburg. Project Funding This project was funded by the Washington State Parks and Recreation Commission. 3 Executive Summary Pacific Biodiversity Institute (PBI) conducted a rare plant and vegetation survey of Riverside State Park (RSP) for the Washington State Parks and Recreation Commission (WSPRC). RSP is located in Spokane County, Washington. A large portion of the park is located within the City of Spokane. RSP extends along both sides of the Spokane River and includes upland areas on the basalt plateau above the river terraces. The park also includes the lower portion of the Little Spokane River and adjacent uplands. The park contains numerous trails, campgrounds and other recreational facilities. The park receives a tremendous amount of recreational use from the nearby population.