Part I Introduction to Series, Methods, and Tolterodine Vs
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Urinary Antispasmodics TCO 02.2018
Therapeutic Class Overview Urinary antispasmodics INTRODUCTION • Overactive bladder (OAB) is defined as urinary urgency, with or without urge incontinence, usually with frequency and nocturia, in the absence of a causative infection or pathological conditions. Urinary incontinence has been shown to greatly reduce quality of life in areas such as mental and general health in addition to physical and social functioning (American Urological Association 2019, Coyne et al 2008, Haab 2014, International Continence Society 2015). Children with OAB usually have detrusor overactivity as diagnosed through cystometric evaluation. Neurogenic detrusor overactivity is predominantly caused by a congenital neural tube defect in children (Austin et al 2016, Franco ○ et al 2020). • Behavioral therapies (eg, bladder training, bladder control strategies, pelvic floor muscle training and fluid management) are considered first-line treatment in all patients with OAB (American Urological Association 2019). • Urinary antispasmodics are used as first-line pharmacological therapy in OAB (American College of Obstetricians and Gynecologists 2015, American Urological Association 2019, Blok et al 2020, Burkhard et al 2018). Anticholinergic therapy has been frequently used in patients with neurogenic detrusor overactivity, but there are limited data in this specific population (Haab 2014). • The○ urinary antispasmodics used for the treatment of OAB belong to 2 classes of drugs, which include anticholinergic compounds known as muscarinic receptor antagonists, and the beta-3 adrenergic agonist (AR), mirabegron. The anticholinergic agents act as antagonists of acetylcholine at muscarinic cholinergic receptors, thereby relaxing smooth muscle in the bladder and decreasing bladder contractions. ○ . Oral immediate-release (IR) and extended-release (ER) formulations (LA, XL, and XR) are available for oxybutynin (Ditropan), tolterodine (Detrol), and trospium. -

Printed Formulary Catalog Basic
Scripps Health Formulary July 2016 Foreword Pharmacy and Therapeutics Committee. MedImpact approves such multi- This document represents the efforts of the MedImpact Healthcare Systems source drugs for addition to the MAC list based on the following criteria: Pharmacy and Therapeutics (P & T) and Formulary Committees to provide physicians A multi-source drug product manufactured by at least one (1) nationally and pharmacists with a method to evaluate the safety, efficacy and cost-effectiveness marketed company. of commercially available drug products. A structured approach to the drug selection At least one (1) of the generic manufacturer’s products must have an “A” process is essential in ensuring continuing patient access to rational drug therapies. rating or the generic product has been determined to be unassociated with The ultimate goal of the Portfolio Formulary is to provide a process and framework to efficacy, safety or bioequivalency concerns by the MedImpact P & T support the dynamic evolution of this document to guide prescribing decisions that Committee. reflect the most current clinical consensus associated with drug therapy decisions. Drug product will be approved for generic substitution by the MedImpact P & T Committee. This is accomplished through the auspices of the MedImpact P & T and Formulary Committees. These committees meet quarterly and more often as warranted to ensure This list is reviewed and updated periodically based on the clinical literature and clinical relevancy of the Formulary. To accommodate changes to this document, pharmacokinetic characteristics of currently available versions of these drug updates are made accessible as necessary. products. As you use this Formulary, you are encouraged to review the information and provide If a member or physician requests a brand name product in lieu of an approved your input and comments to the MedImpact P & T and Formulary Committees. -
![Toviaz) Reference Number: ERX.ST.16 Effective Date: 09.01.17 Last Review Date: 08.17 Line of Business: Commercial [Prescription Drug Plan] Revision Log](https://docslib.b-cdn.net/cover/5292/toviaz-reference-number-erx-st-16-effective-date-09-01-17-last-review-date-08-17-line-of-business-commercial-prescription-drug-plan-revision-log-1025292.webp)
Toviaz) Reference Number: ERX.ST.16 Effective Date: 09.01.17 Last Review Date: 08.17 Line of Business: Commercial [Prescription Drug Plan] Revision Log
Clinical Policy: Fesoterodine (Toviaz) Reference Number: ERX.ST.16 Effective Date: 09.01.17 Last Review Date: 08.17 Line of Business: Commercial [Prescription Drug Plan] Revision Log See Important Reminder at the end of this policy for important regulatory and legal information. Description Fesoterodine (Toviaz®) is a muscarinic antagonist. FDA approved indication Toviaz is indicated for the treatment of overactive bladder with symptoms of urge urinary incontinence, urgency, and frequency. Policy/Criteria Provider must submit documentation (which may include office chart notes and lab results) supporting that member has met all approval criteria It is the policy of health plans affiliated with Envolve Pharmacy Solutions™ that Toviaz is medically necessary when the following criteria are met: I. Initial Approval Criteria A. Step Therapy for Toviaz (must meet all): 1. Age ≥ 18 years; 2. Previous use of 2 formulary generic overactive bladder agents, each trialed for 30 days, unless all are contraindicated or clinically significant adverse effects are experienced; 3. Previous use of Myrbetriq and Vesicare, each trialed for 30 days, unless both are contraindicated or clinically significant adverse effects are experienced; 4. One of the aforementioned trials occurred within the past 6 months, unless all are contraindicated or clinically significant adverse effects are experienced; 5. Dose does not exceed 8 mg per day (1 tablet per day). Approval duration: 12 months II. Continued Therapy A. Step Therapy for Toviaz (must meet all): 1. Currently receiving medication via a health plan affiliated with Envolve Pharmacy Solutions or member has previously met initial approval criteria; 2. If request is for a dose increase, new dose does not exceed 8 mg per day (1 tablet per day). -

Fesoterodine
PATIENT & CAREGIVER EDUCATION Fesoterodine This information from Lexicomp® explains what you need to know about this medication, including what it’s used for, how to take it, its side effects, and when to call your healthcare provider. Brand Names: US Toviaz Brand Names: Canada Toviaz What is this drug used for? It is used to treat an overactive bladder. It is used in some children to treat a bladder problem called neurogenic detrusor overactivity (NDO). What do I need to tell my doctor BEFORE I take this drug? If you are allergic to this drug; any part of this drug; or any other drugs, foods, or substances. Tell your doctor about the allergy and what signs you had. If you have any of these health problems: Glaucoma, liver problems, kidney problems, constipation, slow emptying of the stomach, or trouble passing urine. If you take any other drugs (prescription or OTC, natural products, vitamins). There are many drugs that interact with this drug, like certain drugs that are used for HIV, infections, or seizures. Fesoterodine 1/6 This is not a list of all drugs or health problems that interact with this drug. Tell your doctor and pharmacist about all of your drugs (prescription or OTC, natural products, vitamins) and health problems. You must check to make sure that it is safe for you to take this drug with all of your drugs and health problems. Do not start, stop, or change the dose of any drug without checking with your doctor. What are some things I need to know or do while I take this drug? Tell all of your health care providers that you take this drug. -

Supplemental Table 1: Patient Characteristics, Interventions and Definitions/Variables of Persistence, Adherence and Discontinuation Reported in the Studies
Adherence and persistence to OAB medication Supplemental Table 1: Patient characteristics, interventions and definitions/variables of persistence, adherence and discontinuation reported in the studies. Author (year) and Definition of persistence/adherence/discontinuation and Length of country (database) Participants Drug/intervention* independent variables on persistence follow-up Brostrøm and Hallas n=2477 Any prescription of OAB Patients who continued taking a particular drug for up to 7 years with Up to 7 (2009)1 Male: n=836 (33.8%) medication: no more than 120-day gaps were regarded as experiencing single- years Odense University Female: n=1641 (66.2%) flavoxate (n=21) treatment episodes Pharmacoepidemio- Mean age: 68.3 yearsa oxybutynin TD (n=48) logical Database tolterodine (n=1478) Variables: age, gender, prior use of OAB agents and use of (OPED); Denmark) solifenacin (n=774) anti-diabetic drugs (1999–2006) trospium (n=271) darifenacin (n=52) Chancellor et al (2013)2 n=103 250 First (new) prescription of OAB To be considered a discontinuation, patients were required to have a 2 years IMS Lifelink Database, Male: n≈25 916a (25.1%) medication in adults ≥18 years: gap of at least 45 days in therapy based on fill dates and days’ Connecticut; USA Female: n≈77 334a (74.9%) tolterodine ER (n=43 881)a supply (2005–2008) Mean (SD) age: 58.7 (15.7) solifenacin (n=15 488)a years oxybutynin (n=15 075)a Adherence rate was defined as the proportion of patients filling more darifenacin (n=10 532)a than one prescription with an MPR of ≥80% oxybutynin -

Drug Benefit Council (DBC) Recommendation and Reasons for Recommendation FINAL
Drug Benefit Council (DBC) Recommendation and Reasons for Recommendation FINAL Overactive Bladder Therapeutic Review Description: The purpose of the therapeutic review was to review medications for the treatment of overactive bladder (OAB) in adults. In their review, the DBC considered the following: a systematic review of the clinical evidence, a pharmacoeconomic report; previous CEDAC, CDEC, and DBC Recommendations and Reasons for the applicable agents; Clinical Practice Review from a Specialist, Manufacturer comments; responses to Patient Input Questionnaires from 12 patients; and a Budget Impact Analysis. No patient input was received from caregivers and no patient groups met the inclusion criteria. Dosage Forms: fesoterodine (Toviaz®) 4 and 8 mg tablet; oxybutynin IR (Ditropan®) 5 mg immediate release tablet; oxybutynin ER (Ditropan® XL) 5 and 10 mg extended-release tablet; oxybutynin CR (Uromax®) 10 and 15 mg controlled-release tablet; oxybutynin (Oxytrol™) 36 mg transdermal patch; oxybutynin (Gelnique™) 100 mg/g topical gel; tolterodine (Detrol™) 1 and 2 mg tablet; tolterodine ER (Detrol™ LA) 2 and 4 mg extended-release tablet; solifenacin (Vesicare®) 5 and 10 mg tablet; and darifenacin (Enablex™) 7.5 and 15 mg extended-release tablet. trospium (Trosec®) 20 mg tablet Recommendations: 1. The Drug Benefit Council (DBC) maintains their previous recommendations that oxybutynin CR, oxybutynin gel, and oxybutynin ER not be listed. 2. The DBC recommends that the Ministry of Health (Ministry) should consider adding one of the following long-acting (once daily) agents for patients with OAB who are unable to tolerate oxybutynin IR due to dry mouth. This agent should be selected based on the relative cost of the listed alternatives. -

Pharmacological Treatment of Urinary Incontinence
Committee 8 Pharmacological Treatment of Urinary Incontinence Chairman KARL-ERIK ANDERSSON (USA) Co-Chairman C. R CHAPPLE (U.K) Members L. CARDOZO (U.K), F. C RUZ (Portugal), H. HASHIM (U.K), M.C. MICHEL (The Netherlands), C. TANNENBAUM (Canada), A.J. WEIN (USA) 631 CONTENTS A. INTRODUCTION X. OTHER DRUGS I. PUBLICATION SEARCHES XI. COMBINATIONS II. CENTRAL NERVOUS CONTROL XII. FUTURE POSSIBILITIES III.PERIPHERALNERVOUS CONTROL C.DRUGS USED FOR IV. PATHOGENESIS OF BLADDER TREATMENT OF STRESS CONTROL DISORDERS URINARY INCONTINENCE V. BLADDER CONTRACTION I. α-ADRENCEPTOR AGONISTS VI. MUSCARINIC RECEPTORS II. β-ADRENOCEPTOR ANTAGONISTS B. DRUGS USED FOR TREATMENT OF OVERACTIVE III. β-ADRENOCEPTOR AGONISTS BLADDER SYMPTOMS/ DETRUSOR OVERACTIVITY IV. SEROTONIN-NORADRENALINE UPTAKE INHIBITORS I. ANTIMUSCARINIC (ANTICHOLINERGIC) DRUGS D.DRUGS USED FOR II. DRUGS ACTING ON MEMBRANE TREATMENT OF CHANNELS OVERFLOW INCONTINENCE III. DRUGS WITH “MIXED” ACTION E. HORMONALTREATMENT OF URINARY INCONTINENCE IV. α-ADRENOCEPTOR (AR) ANTAGONISTS I. ESTROGENS V. β-ADRENOCEPTOR AGONISTS II. OTHER STEROID HORMONE VI. PHOSPHODIESTERASE (PDE) RECEPTOR LIGANDS INHIBITORS III. DESMOPRESSIN VII. ANTIDEPRESSANTS VIII. CYCLOOXYGENASE (COX) INHIBITORS F. CONSIDERATIONS IN THE ELDERLY IX. TOXINS REFERENCES 632 Pharmacological Treatment of Urinary Incontinence KARL-ERIK ANDERSSON C. R CHAPPLE, L. CARDOZO, F. CRUZ, H. HASHIM, M.C. MICHEL, C. TANNENBAUM, A.J. WEIN Table 1. ICI assessments 2008: Oxford guidelines (modified) A. INTRODUCTION Levels of evidence Level 1: Systematic reviews, meta-analyses, good The function of the lower urinary tract (LUT) is to store quality randomized controlled clinical trials and periodically release urine, and is dependent on (RCTs) the activity of smooth and striated muscles in the Level 2: RCTs , good quality prospective cohort bladder, urethra, and pelvic floor. -

PREZCOBIX-Pi.Pdf
PREZCOBIX® PREZCOBIX (darunavir and cobicistat) tablets (darunavir and cobicistat) tablets, for oral use • Skin reactions ranging from mild to severe, including Stevens-Johnson Highlights of Prescribing Information Syndrome, toxic epidermal necrolysis, drug rash with eosinophilia and systemic These highlights do not include all the information needed to use PREZCOBIX symptoms and acute generalized exanthematous pustulosis, can occur with safely and effectively. See full prescribing information for PREZCOBIX. PREZCOBIX. Discontinue treatment if severe reaction develops. (5.2) PREZCOBIX® (darunavir and cobicistat) tablets, for oral use • When PREZCOBIX is used in combination with a tenofovir disoproxil fumarate Initial U.S. Approval: 2015 (tenofovir DF) containing regimen, cases of acute renal failure and Fanconi syndrome have been reported. (5.4) --------------------------------RECENT MAJOR CHANGES--------------------------------- • PREZCOBIX is not recommended in combination with other antiretroviral drugs Warnings and Precautions, that require pharmacokinetic boosting. (5.6) Risk of Serious Adverse Reactions or Loss of • Monitor in patients with a known sulfonamide allergy. (5.7) Virologic Response Due to Drug Interactions (5.5) 12/2020 • Patients receiving PREZCOBIX may develop new onset or exacerbations of --------------------------------INDICATIONS AND USAGE--------------------------------- diabetes mellitus/hyperglycemia (5.8), redistribution/accumulation of body fat PREZCOBIX is a two-drug combination of darunavir, a human immunodeficiency -

Toviaz, INN-Fesoterodine Fumarate
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 1 1. NAME OF THE MEDICINAL PRODUCT TOVIAZ 4 mg prolonged-release tablets TOVIAZ 8 mg prolonged-release tablets 2. QUALITATIVE AND QUANTITATIVE COMPOSITION TOVIAZ 4 mg tablets Each prolonged-release tablet contains fesoterodine fumarate 4 mg corresponding to 3.1 mg of fesoterodine. TOVIAZ 8 mg tablets Each prolonged-release tablet contains fesoterodine fumarate 8 mg corresponding to 6.2 mg of fesoterodine. Excipients with known effect TOVIAZ 4 mg tablets Each 4 mg prolonged-release tablet contains 0.525 mg of soya lecithin and 91.125 mg of lactose. TOVIAZ 8 mg tablets Each 8 mg prolonged-release tablet contains 0.525 mg of soya lecithin and 58.125 mg of lactose. For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Prolonged-release tablet. TOVIAZ 4 mg tablets The 4 mg tablets are light blue, oval, biconvex, film-coated, and engraved on one side with the letters ‘FS’. TOVIAZ 8 mg tablets The 8 mg tablets are blue, oval, biconvex, film-coated, and engraved on one side with the letters ‘FT’. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications TOVIAZ is indicated in adults for treatment of the symptoms (increased urinary frequency and/or urgency and/or urgency incontinence) that may occur with overactive bladder syndrome. 2 4.2 Posology and method of administration Posology Adults (including elderly) The recommended starting dose is 4 mg once daily. Based upon individual response, the dose may be increased to 8 mg once daily. The maximum daily dose is 8 mg. Full treatment effect was observed between 2 and 8 weeks. -
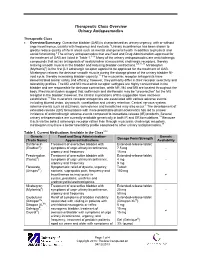
Therapeutic Class Overview Urinary Antispasmodics
Therapeutic Class Overview Urinary Antispasmodics Therapeutic Class • Overview/Summary: Overactive bladder (OAB) is characterized as urinary urgency, with or without urge incontinence, usually with frequency and nocturia.1 Urinary incontinence has been shown to greatly reduce quality of life in areas such as mental and general health in addition to physical and social functioning.2 The urinary antispasmodics that are Food and Drug Administration-approved for the treatment of OAB are listed in Table 1.3-16 Many of the urinary antispasmodics are anticholinergic compounds that act as antagonists of acetylcholine at muscarinic cholinergic receptors, thereby relaxing smooth muscle in the bladder and reducing bladder contractions.3-9,11-16 Mirabegron (Myrbetriq®) is the first β-3 adrenergic receptor agonist to be approved for the treatment of OAB. Mirabegron relaxes the detrusor smooth muscle during the storage phase of the urinary bladder fill- void cycle, thereby increasing bladder capacity.17 The muscarinic receptor antagonists have demonstrated similar safety and efficacy; however, they primarily differ in their receptor selectivity and tolerability profiles. The M2 and M3 muscarinic receptor subtypes are highly concentrated in the bladder and are responsible for detrusor contraction, while M1, M4 and M5 are located throughout the body. Preclinical studies suggest that solifenacin and darifenacin may be “uroselective” for the M3 receptor in the bladder; however, the clinical implications of this suggestion have not been established.18 The muscarinic receptor antagonists are associated with various adverse events including blurred vision, dry mouth, constipation and urinary retention. Central nervous system adverse events such as dizziness, somnolence and headaches may also occur.3 The development of extended-release (ER) formulations with more predictable pharmacokinetics has led to a lower incidence of anticholinergic adverse events compared to immediate-release (IR) products. -

Product Monograph Including Patient Medication Information
PRODUCT MONOGRAPH INCLUDING PATIENT MEDICATION INFORMATION PrTOVIAZ® Fesoterodine fumarate extended-release tablets Extended-release Tablets, 4 mg and 8 mg, Oral Anticholinergic - Antispasmodic Agent Pfizer Canada ULC Date of Initial Authorization: 17,300 Trans-Canada Highway FEB 09, 2012 Kirkland, Quebec H9J 2M5 Date of Revision: JUN 04, 2021 Submission Control Number: 248542 ® C.P. Pharmaceuticals International C.V. Pfizer Canada ULC, Licensee Product Monograph Master Template Template Date: September 2020 Toviaz (fesoterodine fumarate) Page 1 of 37 RECENT MAJOR LABEL CHANGES No major label changes related to safety and efficacy have been made within the past 24 months. TABLE OF CONTENTS Sections or subsections that are not applicable at the time of authorization are not listed. RECENT MAJOR LABEL CHANGES ........................................................................................... 2 TABLE OF CONTENTS ............................................................................................................. 2 PART I: HEALTH PROFESSIONAL INFORMATION ..................................................................... 4 1 INDICATIONS.............................................................................................................. 4 1.1 Pediatrics................................................................................................................... 4 1.2 Geriatrics................................................................................................................... 4 2 CONTRAINDICATIONS................................................................................................ -

A Guide to the Administration of Medicines in the Perioperative
......„„.....NHS Grampian Guideline For The Administration Of Medicines In The Pen-Operative Period Co-ordinators: Consultation Group: Approver: Pre-Operative Assessment See Consultation list Medicine Guidelines and Pharmacist Policies Group Signature: Signature: ...i 0 0 a Identifier: Review Date: Date Approved: NHSG/Guid/Peri0p/ May 2022 May 2019 MGPG1026 Uncontrolled when printed Version 3 Executive Sign-Off This document has been endorsed by the Director of Pharmacy and Medicines Management Signature: Title: Guideline for the Administration of Medicines in the Peri- Operative Period Unique Identifier: NHSG/Guid/PeriOp/MGPG1026 Replaces: NHSG/Guid/PeriOp/MGPG697, Version 2 Across NHS Organisation Directorate Clinical Service Sub Boards Wide Department Area This controlled document shall not be copied in part or whole without the express permission of the author or the author’s representative. Lead Author/Co-ordinator: Pre-Operative Assessment Pharmacist Subject (as per document Clinical Guidance registration categories): Key word(s): Peri-operative medication guidance, gastro intestinal system, cardiovascular system, respiratory, central nervous, infections, endocrine, obstetrics, gynaecology, urinary tract, malignant, nutrition, blood, musculoskeletal, joint, corticosteroid treatment, alternative routes, antiepileptic Process Document: Policy, Guideline Protocol, Procedure or Guideline Document application: NHS Grampian Purpose/description: This policy is designed to be used by pharmacy, nursing and medical staff at pre-assessment