PAX5 Biallelic Genomic Alterations Define a Novel Subgroup of B-Cell
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

GENETICS and GENOMICS Ed
GENETICS AND GENOMICS Ed. Csaba Szalai, PhD GENETICS AND GENOMICS Editor: Csaba Szalai, PhD, university professor Authors: Chapter 1: Valéria László Chapter 2, 3, 4, 6, 7: Sára Tóth Chapter 5: Erna Pap Chapter 8, 9, 10, 11, 12, 13, 14: Csaba Szalai Chapter 15: András Falus and Ferenc Oberfrank Keywords: Mitosis, meiosis, mutations, cytogenetics, epigenetics, Mendelian inheritance, genetics of sex, developmental genetics, stem cell biology, oncogenetics, immunogenetics, human genomics, genomics of complex diseases, genomic methods, population genetics, evolution genetics, pharmacogenomics, nutrigenetics, gene environmental interaction, systems biology, bioethics. Summary The book contains the substance of the lectures and partly of the practices of the subject of ‘Genetics and Genomics’ held in Semmelweis University for medical, pharmacological and dental students. The book does not contain basic genetics and molecular biology, but rather topics from human genetics mainly from medical point of views. Some of the 15 chapters deal with medical genetics, but the chapters also introduce to the basic knowledge of cell division, cytogenetics, epigenetics, developmental genetics, stem cell biology, oncogenetics, immunogenetics, population genetics, evolution genetics, nutrigenetics, and to a relative new subject, the human genomics and its applications for the study of the genomic background of complex diseases, pharmacogenomics and for the investigation of the genome environmental interactions. As genomics belongs to sytems biology, a chapter introduces to basic terms of systems biology, and concentrating on diseases, some examples of the application and utilization of this scientific field are also be shown. The modern human genetics can also be associated with several ethical, social and legal issues. The last chapter of this book deals with these issues. -

Analyses of DNA, RNA and Protein
Analyses of DNA, RNA and Protein What are the early discoveries and technological advances that revolutionized our ability to study human inherited disease? Structure of DNA Central dogma Restriction endonucleases Recombinant DNA technology Cloning Vectors Plasmids Double stranded circular DNA origin of replication selectable marker (antibiotic resistance) 1 or more restriction cutting sites can accommodate DNA fragments 5-10kbp Bacteriophage Lambda Large 25kbp double stranded molecule. Cosmids Can accommodate up to 50kbp of DNA YACs (yeast artificial chromosome Can accommodate up to 1000kbp of DNA BACs (Bacterial artificial chromosome) Can accommodate up to 600 kbp Human genomic plasmid library Also: Phage library YAC library BAC library cDNA library in plasmid Hybridization technology Molecular techniques for Analyzing DNA Southern blotting Detection of gene deletion by Southern blotting Southern blotting can be used to detect large alterations such as Deletions duplications Translocations Point mutations if they alter a restriction enzyme cutting site Quantitative Can be used to analyze large regions of DNA Polymerase Chain Reaction PCR analyses small regions of DNA Sequence of the region must be known to generate primers Not quantitative Advantage of PCR over Southern blotting Fast Sensitive Inexpensive PCR can be used to screen for unknown mutations in small regions of DNA using a variety of approaches, for example: Single strand conformation polymorphism analysis Single strand conformation polymorphism -

Pharmaceutical Compositions Comprising Hydroxychloroquine (HCQ), Curcumin, Piperine/ Bioperine and Uses Thereof in the Medical Field
(19) TZZ 56_8A_T (11) EP 2 561 868 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 27.02.2013 Bulletin 2013/09 A61K 31/12 (2006.01) A61K 31/4706 (2006.01) A61K 45/06 (2006.01) A61P 35/00 (2006.01) (21) Application number: 11178638.0 (22) Date of filing: 24.08.2011 (84) Designated Contracting States: (72) Inventor: Van Oosten, Anton Bernhard AL AT BE BG CH CY CZ DE DK EE ES FI FR GB 3920 Lommel (BE) GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR (74) Representative: DeltaPatents B.V. Designated Extension States: Fellenoord 370 BA ME 5611 ZL Eindhoven (NL) (71) Applicant: Van Oosten, Anton Bernhard 3920 Lommel (BE) (54) Pharmaceutical compositions comprising hydroxychloroquine (HCQ), Curcumin, Piperine/ BioPerine and uses thereof in the medical field (57) The present invention concerns a pharmaceuti- of a premalignant plasma cell proliferative disorder like cal composition containing HCQ, curcumin and BioPer- a monoclonal gammopathy of undetermined significance ine/piperine and its application in the medical field. In (MGUS) and/or smoldering (asymptomatic) multiple my- particular, the composition according to the invention can eloma (SMM) and/or Indolent multiple myeloma (IMM) be advantageously employed in the prevention or treat- and/or to cause remission of a cancer arising from a pre- ment of a subject presenting with a proliferative disorder, malignant plasma cell proliferative disorder like multiple to slow the progression of and/or or to cause regression myeloma (MM). -

High Resolution Molecular Karyotyping and Proteomic Analysis in Hematological Malignancies
Universit`adegli Studi di Catania Scuola Superiore di Catania International PhD in Stem Cells XXIII cycle High resolution molecular karyotyping and proteomic analysis in hematological malignancies Alessandra Romano Coordinator of PhD: Tutor: Prof. Daniele Filippo Condorelli Prof. Francesco di Raimondo Prof. Lance Liotta Prof. Vincenza Barresi A.A. 2007/2010 Stay hard, stay hungry, stay alive: dedicated to all people helped me to work on a dream Abstract In this work we focused on two hematological malignancies to apply the trans- lational meaning of functional genomics: myelodysplastic syndrome (MDS) and its potential evolution to frank acute myeloid leukaemia, and the broad set of monoclonal gammopathies up to multiple myeloma (MM). In both diseases the recent advances obtained thanks to the application of novel therapeutic agents have enlighten the need to target at the same time both neoplastic and sur- rounding microenvironment cells. In MDS we applied the last generation of Affymetrix single nucleotide poly- morphism (SNP)/copy number aberrations (CNA) platform to distinguish so- matic and germline tumor-associated CNAs and loss of heterozigosity (LOHs) to identify possible recurring genomic abnormalities in high risk MDS evolving to AML. In particular in one patient, strictly followed in the clinical evolution from MDS to AML, we were able to define the unique features of the aberrant clone through a bioinformatic-based strategy. For MM, we developed an ex vivo assay to identify signalling associated with differential treatments of fresh bone marrow aspirate samples, confirming the unique constellation of activation in the single patient, and the general trend of a differential behavior among neoplastic and surrounding cells. -
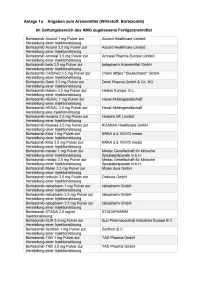
Bortezomib Etc Multiples Myelo
bortezomib AND cyclophosphamide AND dexamethasone AND multi... https://www.ncbi.nlm.nih.gov/pubmed Back to top 11 von 11 16.04.2019, 14:13 4/18/2019 Advanced Search | Cochrane Library https://www.cochranel brary.com/de/advanced-search 2/2 SEARCH QUERY ('bortezomib'/exp OR bortezomib) AND ('cyclophosphamide'/exp OR cyclophosphamide) AND ('dexamethasone'/exp OR dexamethasone) AND ('multiple myeloma'/exp OR 'multiple myeloma') AND 'newly diagnosed' AND 'clinical trial' AND 'induction therapy' RECORD 1 Nicht betrachtet, da bereits in Pubmed-Recherche enthalten Efficacy and Safety of a Weekly Cyclophosphamide-Bortezomib-Dexamethasone Regimen as Induction Therapy Prior to Autologous Stem Cell Transplantation in Japanese Patients with Newly Diagnosed Multiple Myeloma: A Phase 2 Multicenter Trial Tanaka K., Toyota S., Akiyama M., Wakimoto N., Nakamura Y., Najima Y., Doki N., Kakihana K., Igarashi A., Kobayashi T., Ohashi K., Kudo D., Shinagawa A., Takano H., Fujio T., Okoshi Y., Hori M., Kumagai T., Saito T., Mukae J., Yamamoto K., Tsutsumi I., Komeno T., Yoshida C., Yamamoto M., Kojima H. Acta Haematologica (2019) 141:2 (111-118). Date of Publication: 1 Feb 2019 We assessed the efficacy and safety of weekly cyclophosphamide-bortezomib dexamethasone (CBD) induction prior to autologous stem cell transplantation (ASCT) in newly diagnosed Japanese patients with multiple myeloma (MM). This regimen consisted of four 28-day cycles of once-weekly oral cyclophosphamide (300 mg/m(2)), subcutaneous bortezomib (1.3 mg/m(2)), and oral dexamethasone (40 mg). Responding patients underwent stem cell collection followed by ASCT. The primary endpoint was the postinduction rate of achieving a near complete response (nCR) or better. -

Genomic Instability
10/18/16 Genomic Instability Kent Nastiuk, PhD Dept. Cancer Genetics Roswell Park Cancer Institute RPN-530 Oncology for Scientist-I October 18, 2016 Previous lecturers supplying slides/notes/inspiration • Daniel L. Stoler, Ph.D. • Bill Burhans, Ph.D. • Amin Mahpour, (almost Ph.D.?) Genomic Instability RPN-530 Oncology for Scientist-I 2 1 10/18/16 Questions/Outline • What is Genomic instability? • What factors contribute to genome integrity? • How we identify these aberrations? A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns • Notta, et al, Nature October 12, 2016 • doi:10.1038/nature19823 Genomic Instability RPN-530 Oncology for Scientist-I 3 Genomic instability • Cells maintain genome integrity and promote faithful genome propagation by: o Coordinated DNA replication o DNA-damage sensing and repair o Cell-cycle checkpoints • Most checkpoints evolutionarily conserved and are tumor suppressors Genomic Instability RPN-530 Oncology for Scientist-I 4 2 10/18/16 Genomic instability • Drives evolution at the molecular level and generates genetic variation/diversity • Specialized role in generation of variability in developmentally regulated processes o Immunoglobulin diversification • Associated with pathological disorders o Premature aging o Inherited disease o Cancer Genomic Instability RPN-530 Oncology for Scientist-I 5 Cancer • Evolution at a vastly accelerated rate with natural selection favoring the growing tumor mass over the organism. • Successive gene mutations activating oncogenes and inactivating tumor suppressors. Genomic Instability RPN-530 Oncology for Scientist-I 6 3 10/18/16 7 Genomic Instability RPN-530 Oncology for Scientist-I Early Molecular Model of Tumor Progression - Vogelstein • Hypothesis: Mutation in one gene associated with each step in progression. -

CYTOGENETIC BIOINFORMATICS of CHROMOSOMAL ABERRATIONS and GENETIC DISORDERS: DATA-MINING of RELEVANT BIOSTATISTICAL FEATURES By
CYTOGENETIC BIOINFORMATICS OF CHROMOSOMAL ABERRATIONS AND GENETIC DISORDERS: DATA-MINING OF RELEVANT BIOSTATISTICAL FEATURES by Jagadeshwari Karri A Thesis Submitted to the Faculty of the College of Engineering and Computer Science in Partial Fulfillment of the Requirements for the Degree of Master of Science Florida Atlantic University Boca Raton, Florida December 2012 ACKNOWLEDGMENTS This study would not have been completed without the direction and guidance from Dr. Perambur S. Neelakanta, the Committee Chairperson and thesis advisor. Dr. Neelakanta taught me how to be patient as well as he was available to provide constructive feedback regarding questions related to research, algorithms and computations. My sincere thanks are extended to him for his time and editing this thesis. I wish to thank the Committee Members, Drs. Abhijit Pandya, Mirjana Pavlovic and Dolores DeGroff for their time serving as members of the committee and providing their educated assessment to this thesis. Finally, I would like to greatly thankful to my parents, my in-law and my husband and son for their moral support, constant encouragement and enormous patience while preparing this thesis and especially for all those years of pursuing my education. iii ABSTRACT Author: Jagadeshwari Karri Title: Cytogenetic Bioinformatics of Chromosomal Aberrations and Genetic Disorders: Data-mining of Relevant Biostatistical Features Institution: Florida Atlantic University Thesis Advisor: Dr. Perambur S. Neelakanta Degree: Master of Science Year 2012 Cytogenetics is a study on the genetic considerations associated with structural and functional aspects of the cells with reference to chromosomal inclusions. Chromosomes are structures within the cells containing body’s information in the form of strings of DNA. -

Present and Future Salmonid Cytogenetics
G C A T T A C G G C A T genes Article Present and Future Salmonid Cytogenetics Muhammet Gaffaroglu 1 , Zuzana Majtánová 2 , Radka Symonová 3,* ,Šárka Pelikánová 2, Sevgi Unal 4 , ZdenˇekLajbner 5 and Petr Ráb 2 1 Department of Molecular Biology and Genetics, Faculty of Science, University of Ahi Evran, Kirsehir 40200, Turkey; mgaff[email protected] 2 Laboratory of Fish Genetics, Institute of Animal Physiology and Genetics, Czech Academy of Sciences, 27721 Libˇechov, Czech Republic; [email protected] (Z.M.); [email protected] (Š.P.); [email protected] (P.R.) 3 Department of Bioinformatics, Wissenschaftszentrum Weihenstephan, Technische Universität München, 85354 Freising, Germany 4 Department of Molecular Biology and Genetics, Faculty of Science, Bartin University, Bartin 74000, Turkey; [email protected] 5 Physics and Biology Unit, Okinawa Institute of Science and Technology, Graduate University, Onna, Okinawa 904 0495, Japan; [email protected] * Correspondence: [email protected] or [email protected] Received: 6 November 2020; Accepted: 2 December 2020; Published: 6 December 2020 Abstract: Salmonids are extremely important economically and scientifically; therefore, dynamic developments in their research have occurred and will continue occurring in the future. At the same time, their complex phylogeny and taxonomy are challenging for traditional approaches in research. Here, we first provide discoveries regarding the hitherto completely unknown cytogenetic characteristics of the Anatolian endemic flathead trout, Salmo platycephalus, and summarize the presently known, albeit highly complicated, situation in the genus Salmo. Secondly, by outlining future directions of salmonid cytogenomics, we have produced a prototypical virtual karyotype of Salmo trutta, the closest relative of S. -

Clear Cell Papillary Renal Cell Carcinoma: Molecular Profile and Virtual Karyotype
Human Pathology (2019) 91,52–60 www.elsevier.com/locate/humpath Original contribution Clear cell papillary renal cell carcinoma: molecular profile and virtual karyotype☆,☆☆ Diana M. Morlote, MD a,⁎,1, Shuko Harada, MD a,1, Denise Batista, PhD b, Jennifer Gordetsky, MD c,1, Soroush Rais-Bahrami, MD d,e,1 aDepartment of Pathology, University of Alabama at Birmingham, Birmingham, AL 35233, USA bJohns Hopkins University School of Medicine, Baltimore, MD 21205, USA cDepartment of Pathology, Vanderbilt University Medical Center, Nashville, TN 37232, USA dDepartment of Urology, University of Alabama at Birmingham, Birmingham, AL 35233, USA eDepartment of Radiology, University of Alabama at Birmingham, Birmingham, AL 35233, USA Received 15 March 2019; revised 26 May 2019; accepted 31 May 2019 Keywords: Summary Clear cell papillary renal cell carcinoma (CCP-RCC) is a recently recognized tumor that shares Clear cell papillary renal morphologic features of both clear cell renal cell carcinoma and papillary renal cell carcinoma but be- cell carcinoma; haves in a more indolent fashion. To date, there is little molecular information available on CCP-RCC. Genetic profile; DNA was extracted from formalin-fixed, paraffin-embedded tissue blocks of 22 cases of CCP-RCC at Molecular study; the University of Alabama at Birmingham. Targeted next-generation sequencing and single-nucleotide VHL mutations; polymorphism array were performed on all cases. Next-generation sequencing analysis found 30 somatic Virtual karyotyping; variants across 63.3% of cases. Seventeen variants (56.7%) were predicted to be deleterious or possibly/ Low-grade renal cell probably damaging. Single-nucleotide polymorphism array analysis found copy number abnormalities carcinoma and/or loss of heterozygosity in 22.7% of cases. -

Insight on Pathogen Defense Mechanisms in the Sugarcane Transcriptome
® Functional Plant Science and Biotechnology ©2012 Global Science Books Insight on Pathogen Defense Mechanisms in the Sugarcane Transcriptome Ana Carolina Wanderley-Nogueira1 • Ederson Akio Kido1 • Nina da Mota Soares-Cavalcanti1 • Luis Carlos Belarmino1 • João Pacífico Bezerra-Neto1 • William Lee Burnquist2 • Sabrina Moutinho Chabregas2 • José Ivo Baldani3 • Ana Maria Benko-Iseppon1* 1 Genetics Department, Center of Biological Sciences, Universidade Federal de Pernambuco, Av. Prof. Morais Rego 1235, CEP 50.670-420, Recife, PE, Brazil 2 Centro de Tecnologia Canavieira (CTC), Bairro Santo Antonio, CEP 13400-970, Piracicaba, SP, Brazil 3 Embrapa Agrobiologia (CNPAB), Rodovia BR 465 km 7, CEP 23890-000, Seropédica, RJ, Brazil Corresponding author : * [email protected] ABSTRACT Recognition of pathogen and activation of defense mechanisms is a common feature known from all multicellular organisms. Among higher plants, systemic acquired resistance (SAR) is known to activate pathogenesis-related (PR) genes after recognition of the pathogen mediated by a resistance (R) gene. Both gene classes (R and PR) represent the main mechanism against biotic and sometimes also abiotic stresses. Therefore, the identification of SAR metabolism components is an important concern regarding plant breeding. Sugarcane (Saccharum officinarum) is a major tropical and subtropical crop, grown in more than 120 countries, being especially important due to its value for the production of bioethanol, constituting a renewable energy source, besides the sugar production. The present work brings an overview on sugarcane resistance and pathogenesis-related genes, regarding their structure, abundance and role in the plant-pathogen metabolic pathway and also regarding their distribution, as compared with rice. For this purpose a collection of 282,818 expressed sequences tags have been evaluated to identify R and PR genes as well as important factors identified in the classic plant-pathogen metabolic KEGG pathway using rice full length cDNA as seed-sequences. -

SEETHRAM Prenatal Screening
27/07/2017 Prenatal Screening All that you need to know….. Fertility Symposium Ken Seethram, MD, FRCSC, FACOG Pacific Centre for Reproductive Medicine Clinical Assistant Professor, UBC & University of Alberta [email protected] Outline • To understand • Prenatal Screening and amniocentesis/CVS • Non-Invasive Prenatal Testing/Screening • To understand the scope of NIPT and its variations • To understand the detection rates and limitations of NIPT • To understand the various forms of prenatal screening, and who gets what in BC 1 27/07/2017 Disclosure • I have no financial interest in the current NIPT providers – we receive no research grants, educational grants, or other initiatives from any NIPT provider • We draw and send plasma on patients for NIPT in our centre • We perform first trimester screening (non-insured service) in our centre • Warning: trade names will be used during this talk Prenatal Screening & Diagnosis for Chromosome Abnormalities • T21 is one of the most common aneuploidy to affect live-born children and has a background prevalence of 1:691, increasing with maternal age • What causes T21 – hypo-methylation by the extra chromosome • Prenatal diagnosis relies upon the procurement of fetal cells via amniocentesis (ACT) / trophoblast via chorionic villus sampling (CVS) 2 27/07/2017 1977-That would have been the end of this talk Prenatal Screening & Diagnosis for Chromosome Abnormalities • Simpler times back then; if your age based risk exceeded the risk of diagnostic testing, then do the test (which was roughly age 35-38) • So the only screening tool we had was age • What is the risk of Amniocentesis or CVS? • For the last 40 years, it has been quoted at 1% 3 27/07/2017 What is the RISK of CVS/ACT • A recent meta-analysis (Akolekar, et al (2015) Procedure-related risk of miscarriage following amniocentesis and chorionic villus sampling: a systematic review and meta-analysis. -

Genetics and Genomics
GENETICS AND GENOMICS Editor: Csaba Szalai, PhD, university professor Authors: Chapter 1: Valéria László Chapter 2, 3, 4, 6, 7: Sára Tóth Chapter 5: Erna Pap Chapter 8, 9, 10, 11, 12, 13, 14: Csaba Szalai Chapter 15: András Falus and Ferenc Oberfrank Keywords: Mitosis, meiosis, mutations, cytogenetics, epigenetics, Mendelian inheritance, genetics of sex, developmental genetics, stem cell biology, oncogenetics, immunogenetics, human genomics, genomics of complex diseases, genomic methods, population genetics, evolution genetics, pharmacogenomics, nutrigenetics, gene environmental interaction, systems biology, bioethics. Summary The book contains the substance of the lectures and partly of the practices of the subject of ‘Genetics and Genomics’ held in Semmelweis University for medical, pharmacological and dental students. The book does not contain basic genetics and molecular biology, but rather topics from human genetics mainly from medical point of views. Some of the 15 chapters deal with medical genetics, but the chapters also introduce to the basic knowledge of cell division, cytogenetics, epigenetics, developmental genetics, stem cell biology, oncogenetics, immunogenetics, population genetics, evolution genetics, nutrigenetics, and to a relative new subject, the human genomics and its applications for the study of the genomic background of complex diseases, pharmacogenomics and for the investigation of the genome environmental interactions. As genomics belongs to sytems biology, a chapter introduces to basic terms of systems biology, and concentrating on diseases, some examples of the application and utilization of this scientific field are also be shown. The modern human genetics can also be associated with several ethical, social and legal issues. The last chapter of this book deals with these issues.