Annex I Summary of Product Characteristics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Dilantin (Phenytoin Sodium) Extended Oral Capsule Three Times Daily and the Dosage Then Adjusted to Suit Individual Requirements
Dilantin® (Phenytoin Sodium) 100 mg Extended Oral Capsule DESCRIPTION Phenytoin sodium is an antiepileptic drug. Phenytoin sodium is related to the barbiturates in chemical structure, but has a five-membered ring. The chemical name is sodium 5,5-diphenyl-2, 4-imidazolidinedione, having the following structural formula: Each Dilantin— 100 mg Extended Oral Capsule—contains 100 mg phenytoin sodium. Also contains lactose monohydrate, NF; confectioner’s sugar, NF; talc, USP; and magnesium stearate, NF. The capsule body contains titanium dioxide, USP and gelatin, NF. The capsule cap contains FD&C red No. 28; FD&C yellow No. 6; and gelatin NF. Product in vivo performance is characterized by a slow and extended rate of absorption with peak blood concentrations expected in 4 to 12 hours as contrasted to Prompt Phenytoin Sodium Capsules, USP with a rapid rate of absorption with peak blood concentration expected in 1½ to 3 hours. CLINICAL PHARMACOLOGY Phenytoin is an antiepileptic drug which can be used in the treatment of epilepsy. The primary site of action appears to be the motor cortex where spread of seizure activity is inhibited. Possibly by promoting sodium efflux from neurons, phenytoin tends to stabilize the threshold against hyperexcitability caused by excessive stimulation or environmental changes capable of reducing membrane sodium gradient. This includes the reduction of posttetanic potentiation at synapses. Loss of posttetanic potentiation prevents cortical seizure foci from detonating adjacent cortical areas. Phenytoin reduces the maximal activity of brain stem centers responsible for the tonic phase of tonic-clonic (grand mal) seizures. The plasma half-life in man after oral administration of phenytoin averages 22 hours, with a range of 7 to 42 hours. -

THE SPECIFICITY of DRUG BINDING SITES on HUMAN SERUM ALBUMIN Ingvar Sjòholm
THE SPECIFICITY OF DRUG BINDING SITES ON HUMAN SERUM ALBUMIN Ingvar Sjòholm Today, it is well established that the binding of drugs in serum will strongly influence the pharmacokinetic parameters of a drug, such as its distribution volume and clearance. It is also evident that the binding of the drug—in serum and elsewhere in the tissues—will have an influence on the duration and intensity of the pharmacological effect. Several excellent papers and reviews have dealt with these issues in recent years.1"** It is obvious that albumin, being the most abundant protein species in the extracellular fluids, is the most im- portant drug-binding protein, although other proteins can play a pharmacokinetic role. Thus, e.g., orosomucoid (aj-acid glyco- protein) can bind some basic and neutral drugs ,9 and lipoproteins some highly hydrophobic drugs.10 The primary structure of human serum albumin (HSA) is now known.1:L'^2 However, all efforts to study the three-dimensional structure by x-ray spectroscopy have hitherto failed, and a detailed knowledge of the mechanisms involved in the binding of drugs or endogenous compounds is still missing. The broad binding specificity of HSA is remarkable. Several compounds of widely different struc- ture can be bound with high affinity—e.g., fatty acids, bilirubin, tryptophan, as well as many drugs. It is also striking that different reports from quantitative studies on the binding of different com- pounds have shown varying results, which cannot be solely explained by technical problems or different experimental conditions. All avail- able information indicates that HSA is a highly "flexible" and "adapt- able" molecule, the structure of which can be strongly influenced by different "modulating" substances. -

NINDS Custom Collection II
ACACETIN ACEBUTOLOL HYDROCHLORIDE ACECLIDINE HYDROCHLORIDE ACEMETACIN ACETAMINOPHEN ACETAMINOSALOL ACETANILIDE ACETARSOL ACETAZOLAMIDE ACETOHYDROXAMIC ACID ACETRIAZOIC ACID ACETYL TYROSINE ETHYL ESTER ACETYLCARNITINE ACETYLCHOLINE ACETYLCYSTEINE ACETYLGLUCOSAMINE ACETYLGLUTAMIC ACID ACETYL-L-LEUCINE ACETYLPHENYLALANINE ACETYLSEROTONIN ACETYLTRYPTOPHAN ACEXAMIC ACID ACIVICIN ACLACINOMYCIN A1 ACONITINE ACRIFLAVINIUM HYDROCHLORIDE ACRISORCIN ACTINONIN ACYCLOVIR ADENOSINE PHOSPHATE ADENOSINE ADRENALINE BITARTRATE AESCULIN AJMALINE AKLAVINE HYDROCHLORIDE ALANYL-dl-LEUCINE ALANYL-dl-PHENYLALANINE ALAPROCLATE ALBENDAZOLE ALBUTEROL ALEXIDINE HYDROCHLORIDE ALLANTOIN ALLOPURINOL ALMOTRIPTAN ALOIN ALPRENOLOL ALTRETAMINE ALVERINE CITRATE AMANTADINE HYDROCHLORIDE AMBROXOL HYDROCHLORIDE AMCINONIDE AMIKACIN SULFATE AMILORIDE HYDROCHLORIDE 3-AMINOBENZAMIDE gamma-AMINOBUTYRIC ACID AMINOCAPROIC ACID N- (2-AMINOETHYL)-4-CHLOROBENZAMIDE (RO-16-6491) AMINOGLUTETHIMIDE AMINOHIPPURIC ACID AMINOHYDROXYBUTYRIC ACID AMINOLEVULINIC ACID HYDROCHLORIDE AMINOPHENAZONE 3-AMINOPROPANESULPHONIC ACID AMINOPYRIDINE 9-AMINO-1,2,3,4-TETRAHYDROACRIDINE HYDROCHLORIDE AMINOTHIAZOLE AMIODARONE HYDROCHLORIDE AMIPRILOSE AMITRIPTYLINE HYDROCHLORIDE AMLODIPINE BESYLATE AMODIAQUINE DIHYDROCHLORIDE AMOXEPINE AMOXICILLIN AMPICILLIN SODIUM AMPROLIUM AMRINONE AMYGDALIN ANABASAMINE HYDROCHLORIDE ANABASINE HYDROCHLORIDE ANCITABINE HYDROCHLORIDE ANDROSTERONE SODIUM SULFATE ANIRACETAM ANISINDIONE ANISODAMINE ANISOMYCIN ANTAZOLINE PHOSPHATE ANTHRALIN ANTIMYCIN A (A1 shown) ANTIPYRINE APHYLLIC -

Decreased Serum Concentrations of Tamoxifen and Its Metabolites Induced by Aminoglutethimide1
(CANCER RESEARCH 50. 5851-5857. September 15. 1990) Decreased Serum Concentrations of Tamoxifen and Its Metabolites Induced by Aminoglutethimide1 Ernst A. Lien,2 Gun Anker, Per Eystein Lgnning, Einar Solheim, and Per M. Ueland Department i>j'Pharmacology and Toxicology [E. A. I... E. S]; Clinical Pharmacology I nit. Department of Pharmacology and Toxicology /P. M. l './; and Department of Oncology I(i. A., P. fi. Lj, L'niversity of Bergen, .\-502l, Bergen, \onvay ABSTRACT Aminoglutethimide inhibits the enzyme aromatase, which converts androgens to estrogens in peripheral fat tissue (3). The anticstrogen tamoxifen and the aromatase inhibitor aminoglute- This conversion is the main estrogen source in postmenopausal thimide show similar response rates when used in the endocrine manage women. In addition, aminoglutethimide may reduce the con ment of advanced breast cancer. However, numerous clinical trials have centration of plasma estrogens by enhancement of estrogen demonstrated no increase in response rate from treatment with the drug combination of tamoxifen plus aminoglutethimide. We investigated the metabolism (10, 11). Aminoglutethimide causes response rates possibility of a pharmacokinetic interaction between these two drugs in in postmenopausal breast cancer patients similar to those of six menopausa! woman with breast cancer. All patients were investigated tamoxifen, but because of more frequent side effects aminoglu under three different conditions (termed phases A, B, and C). The steady tethimide is generally used after tamoxifen as a second line state kinetics of tamoxifen were determined when administered alone endocrine treatment (12). (phase A) and after coadministration of aminoglutethimide for 6 weeks Combination therapy with tamoxifen plus aminoglutethi (phase B). -

Adverse Effects of Digoxin, As Xenoestrogen, on Some Hormonal and Biochemical Patterns of Male Albino Rats Eman G.E.Helal *, Mohamed M.M
The Egyptian Journal of Hospital Medicine (October 2013) Vol. 53, Page 837– 845 Adverse Effects of Digoxin, as Xenoestrogen, on Some Hormonal and Biochemical Patterns of Male Albino Rats Eman G.E.Helal *, Mohamed M.M. Badawi **,Maha G. Soliman*, Hany Nady Yousef *** , Nadia A. Abdel-Kawi*, Nashwa M. G. Abozaid** Department of Zoology, Faculty of Science, Al-Azhar University (Girls)*, Department of Biochemistry, National organization for Drug Control and Research** Department of Biological and Geological Sciences, Faculty of Education, Ain Shams University*** Abstract Background: Xenoestrogens are widely used environmental chemicals that have recently been under scrutiny because of their possible role as endocrine disrupters. Among them is digoxin that is commonly used in the treatment of heart failure and atrial dysrhythmias. Digoxin is a cardiac glycoside derived from the foxglove plant, Digitalis lanata and suspected to act as estrogen in living organisms. Aim of the work: The purpose of the current study was to elucidate the sexual hormonal and biochemical patterns of male albino rats under the effect of digoxin treatment. Material and Methods: Forty six male albino rats (100-120g) were divided into three groups (16 rats for each). Half of the groups were treated daily for 15 days and the other half for 30 days. Control group: Animals without any treatment. Digoxin L group: orally received digoxin at low dose equivalent of 0.0045mg/200g.b.wt. Digoxin H group: administered digoxin orally at high dose equivalent of 0.0135mg/200g.b.wt. At the end of the experimental periods, blood was collected and serum was separated for estimation the levels of prolactin (PRL), FSH, LH, total testosterone (total T), aspartate amino transferase (AST), alanine amino transferase (ALT), alkaline phosphatase (ALP), urea, creatinine, total proteins, albumin, total lipids, total cholesterol (total-chol), Triglycerides (TG), low density lipoprotein cholesterol (LDL-chol) and high density lipoprotein cholesterol (HDL-chol). -

A Critical Appraisal of a Further Three New Commercial Digoxin Radioimmunoassay Kits with Reference to Cross-Reacting Substances
Wood and Wächter: Appraisal of three digoxin radioimmunoassay kits 77 J. Clin. Chern. Clin. Biochem. Vol. 17,1979, pp. 77-83 A Critical Appraisal of a Further Three New Commercial Digoxin Radioimmunoassay Kits with Reference to Cross-Reacting Substances By W. G. Wood and Christine Wächter Laboratorien für Klinische Chemie und Endokrinologie, Medizinische Klinik Innenstadt der Universität München (Received July 20/September 11, 1978) Summary: A further 3 digoxin radioimmunoassay (RIA) kits have been evaluated for performance and cross-reaction with digitoxin, spironolactone, canrenone and furosemide (Lasix-Hoechst). Effects of serum protein concentrations have also been tested. The kits tested were from the following manufacturers: A) Diagnostic Products Corporation Digoxin RIA Kit. B) Byk-Mallinckrodt SPAC Digoxin Kit. C) Boehringer-Mannheim Digoxin RIA Kit. All kits used a 125I-labelled tracer. Kit A used a conventional liquid phase system using double-antibody separation for bound and free drug, Kits B and C used a solid-phase antibody coated tube method. All kits showed a lower cross-reaction to digitoxin than quoted by the manufacturer. Cross-reaction to spironolactone (Aldactone — Boehringer-Mannheim) was less than 1.50 nmol/I at a serum concentra- tion of 125 mg/1 Aldactone in all 3 kits. The cross-reaction to canrenone was somewhat higher, 5.2 nmol/1 "digoxin" being measured in one kit at a serum canrenone concentration of 125 mg/1. There was no cross-reaction with furo- semide in any kit, even at ä serum concentration of 5 g/1. The coated-tube assays were affected by serum albumin and globulin concentration changes, one kit showing a difference of over 50% binding in the range 1—20% albumin. -

Study Protocol, Statistical Analysis Plan, And
Amendment Protocol Number: 04C0257-Q Reference Number: 366512 Principal Investigator: Ravi Madan NCI GMB 301.480.7168 [email protected] (NIH Employee Name, Institute/Branch, Telephone and e-mail) Protocol Title: A Phase II Trial of Docetaxel, Thalidomide, Prednisone, and Bevacizumab in Patients with Androgen-Independent Prostate Cancer SIGNATURES Principal Investigator (*): Ravi Madan - applied signature on 02/09/2017 9:47 AM EST Accountable Investigator: PI is the Accountable Investigator Branch Chief/CC Department Head (**): James L Gulley, MD PhD - applied signature on 02/06/2017 3:53 PM EST Medical Advisory Investigator (if applicable): N/A Lead Associate Investigator signature: N/A Referral Contact signatures: N/A Associate Investigators signatures: N/A For Institute/Center Scientific Review Committee: N/A Other IC Clinical Director signatures: N/A APPROVALS IRB Chair: Michael Hamilton - applied signature on 03/07/2017 7:58 PM EST Clinical Director: N/A CONCURRENCE OPS Protocol Specialist: Royal Reed AM Q 03/17/17 Signature Print Name Date * Signature signifies that investigators on this protocol have been informed that the collection and use of personally identifiable information at the NIH are maintained in a system of record governed under provisions of the Privacy Act of 1974. The information provided is mandatory for employees of the NIH to perform their assigned duties as related to the administration and reporting of intramural research protocols and used solely for those purposes. Questions may be addressed to the Protrak System Owner. ** I have reviewed this research project and considered the NIH Policy for Inclusion of Women and Minorities in Clinical Research. -

EUROPEAN PHARMACOPOEIA 10.0 Index 1. General Notices
EUROPEAN PHARMACOPOEIA 10.0 Index 1. General notices......................................................................... 3 2.2.66. Detection and measurement of radioactivity........... 119 2.1. Apparatus ............................................................................. 15 2.2.7. Optical rotation................................................................ 26 2.1.1. Droppers ........................................................................... 15 2.2.8. Viscosity ............................................................................ 27 2.1.2. Comparative table of porosity of sintered-glass filters.. 15 2.2.9. Capillary viscometer method ......................................... 27 2.1.3. Ultraviolet ray lamps for analytical purposes............... 15 2.3. Identification...................................................................... 129 2.1.4. Sieves ................................................................................. 16 2.3.1. Identification reactions of ions and functional 2.1.5. Tubes for comparative tests ............................................ 17 groups ...................................................................................... 129 2.1.6. Gas detector tubes............................................................ 17 2.3.2. Identification of fatty oils by thin-layer 2.2. Physical and physico-chemical methods.......................... 21 chromatography...................................................................... 132 2.2.1. Clarity and degree of opalescence of -
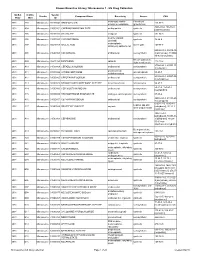
Known Bioactive Library: Microsource 1 - US Drug Collection
Known Bioactive Library: Microsource 1 - US Drug Collection ICCB-L ICCB-L Vendor Vendor Compound Name Bioactivity Source CAS Plate Well ID antifungal, inhibits Penicillium 2091 A03 Microsource 00200046 GRISEOFULVIN 126-07-8 mitosis in metaphase griseofulvum 3505-38-2, 486-16-8 2091 A04 Microsource 01500161 CARBINOXAMINE MALEATE antihistaminic synthetic [carbinoxamine] 2091 A05 Microsource 00200331 SALSALATE analgesic synthetic 552-94-3 muscle relaxant 2091 A06 Microsource 01500162 CARISOPRODOL synthetic 78-44-4 (skeletal) antineoplastic, 2091 A07 Microsource 00210369 GALLIC ACID insect galls 149-91-7 astringent, antibacterial 66592-87-8, 50370-12- 2091 A08 Microsource 01500163 CEFADROXIL antibacterial semisynthetic 2 [anhydrous], 119922- 89-9 [hemihydrate] Rheum palmatum, 2091 A09 Microsource 00211468 DANTHRON cathartic 117-10-2 Xyris semifuscata 27164-46-1, 25953-19- 2091 A10 Microsource 01500164 CEFAZOLIN SODIUM antibacterial semisynthetic 9 [cefazolin] glucocorticoid, 2091 A11 Microsource 00300024 HYDROCORTISONE adrenal glands 50-23-7 antiinflammatory 64485-93-4, 63527-52- 2091 A12 Microsource 01500165 CEFOTAXIME SODIUM antibacterial semisynthetic 6 [cefotaxime] 2091 A13 Microsource 00300029 DESOXYCORTICOSTERONE ACETATE mineralocorticoid adrenocortex 56-47-3 58-71-9, 153-61-7 2091 A14 Microsource 01500166 CEPHALOTHIN SODIUM antibacterial semisynthetic [cephalothin] 2091 A15 Microsource 00300034 TESTOSTERONE PROPIONATE androgen, antineoplastic semisynthetic 57-85-2 24356-60-3, 21593-23- 2091 A16 Microsource 01500167 CEPHAPIRIN SODIUM -

Digitalis Toxicity Randy Easterling, M.D., and Paul E
J Am Board Fam Pract: first published as 10.3122/jabfm.2.1.49 on 1 January 1989. Downloaded from Digitalis Toxicity Randy Easterling, M.D., and Paul E. Tietze, M.D. Abstract: Digitalis toxicity is a frequently encoun bodies represents a significant advance in the treat tered clinical problem. Drug interactions leading to ment of this problem and can be lifesaving in digitalis toxicity are very common. Within this the setting of massive overdose. A case report of digi group, the interaction of digitalis and verapamil is talis toxicity and a review of this problem and its increasingly recognized as an important \:ause of treatment are presented. (J Am Bd Fam Pract 1989; digitalis toxicity. The use of digoxin-binding anti- 2:49-54.) Digitalis preparations are frequently prescribed, ble and had miotic pupils. The physical examination and a significant number of patients, especially was otherwise unremarkable. the elderly, take them regularly. Because of the A 12-lead electrocardiogram showed atrial fi ubiquitous use of this drug and the increased fre brillation with a high-grade delay in AV conduc quency of digitalis toxicity, the rapid diagnosis of tion. The Q-T interval was markedly shortened. digitalis toxicity is essential. Results from measur The ventricular rate was 31, and there were long ing digitalis blood levels can help the physician asystolic intervals in V4-V6. There was sagging of provide optimal care to patients. Where digitalis the SoT segments in leads II, III, and aVF and up levels are not available, the physician must de ward sloping SoT segments in aVR. -

Early Refill Drug Utilization Review Drugs
Early Refill Drug Utilization Review Drugs Published: 05/01/2012 Point-of-Sale Drug Authorization and Policy Drug Name Effective Date Override Override Center Override Acarbose X 11/8/2008 Acebutolol hydrochloride X 11/8/2008 acetaminophen X 11/8/2008 acetazolamide X 11/8/2008 acetazolamide sodium X 11/8/2008 acetylcholine X 11/8/2008 acetylcysteine X 11/8/2008 Acitretin X 11/8/2008 Acrivastine X 11/8/2008 activated charcoal X 11/8/2008 adalimumab X 11/8/2008 Adapalene X 11/8/2008 Adenosine X 11/8/2008 albumin human X 11/8/2008 Albuterol X 11/8/2008 albuterol sulfate X 11/8/2008 alclometasone dipropionate X 11/8/2008 aldesleukin X 11/8/2008 alendronate sodium X 11/8/2008 alfentanil hydrochloride X 11/8/2008 aliskiren hemifumarate X 11/8/2008 Alitretinoin X 11/8/2008 almotriptan malate X 11/8/2008 alosetron hydrochloride X 11/8/2008 Alprazolam X 1/6/2010 Aluminum X 11/8/2008 aluminum sulfate X 11/8/2008 amantadine hydrochloride X 11/8/2008 amcinonide X 11/8/2008 amino acids X 11/8/2008 aminophylline X 11/8/2008 amiodarone hydrochloride X 11/8/2008 amitriptyline hydrochloride X 11/8/2008 amlodipine besylate X 11/8/2008 Ammonium X 11/8/2008 Ammonium chloride X 11/8/2008 amobarbital X 1/6/2010 Point-of-Sale Drug Authorization and Policy Drug Name Effective Date Override Override Center Override Amoxicillin trihydrate X 11/8/2008 amphetamine X 1/6/2010 amyl nitrite X 11/8/2008 Amylase X 11/8/2008 apraclonidine hydrochloride X 11/8/2008 arformoterol tartrate X 11/8/2008 Arginine X 11/8/2008 arginine hydrochloride X 11/8/2008 aripiprazole X -

Guidelines for Treatment of Drug-Susceptible Tuberculosis and Patient Care
TREATMENT OF TUBERCULOSIS Guidelines for treatment of drug-susceptible tuberculosis and patient care 2017 UPDATE Annex 6 ESSENTIAL FIRST-LINE ANTITUBERCULOSIS DRUGS TREATMENT OF TUBERCULOSIS Guidelines for treatment of drug-susceptible tuberculosis and patient care 2017 UPDATE Annex 6 ESSENTIAL FIRST-LINE ANTITUBERCULOSIS DRUGS Guidelines for treatment of drug-susceptible tuberculosis and patient care, 2017 update Contents: Web Annex 3: GRADE evidence profiles – Web Annex 4: Evidence-to-decision tables – Annex 5: Reports of the systematic reviews – Annex 6: Essential first-line antituberculosis drugs ISBN 978-92-4-155000-0 © World Health Organization 2017 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization.