Synthesis and Structural Characterisation of Bismuth(III) Hydroxamates and Their Activity Against Helicobacter Pylori
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Alternative Oxidase: a Mitochondrial Respiratory Pathway to Maintain Metabolic and Signaling Homeostasis During Abiotic and Biotic Stress in Plants
Int. J. Mol. Sci. 2013, 14, 6805-6847; doi:10.3390/ijms14046805 OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Review Alternative Oxidase: A Mitochondrial Respiratory Pathway to Maintain Metabolic and Signaling Homeostasis during Abiotic and Biotic Stress in Plants Greg C. Vanlerberghe Department of Biological Sciences and Department of Cell and Systems Biology, University of Toronto Scarborough, 1265 Military Trail, Toronto, ON, M1C1A4, Canada; E-Mail: [email protected]; Tel.: +1-416-208-2742; Fax: +1-416-287-7676 Received: 16 February 2013; in revised form: 8 March 2013 / Accepted: 12 March 2013 / Published: 26 March 2013 Abstract: Alternative oxidase (AOX) is a non-energy conserving terminal oxidase in the plant mitochondrial electron transport chain. While respiratory carbon oxidation pathways, electron transport, and ATP turnover are tightly coupled processes, AOX provides a means to relax this coupling, thus providing a degree of metabolic homeostasis to carbon and energy metabolism. Beside their role in primary metabolism, plant mitochondria also act as “signaling organelles”, able to influence processes such as nuclear gene expression. AOX activity can control the level of potential mitochondrial signaling molecules such as superoxide, nitric oxide and important redox couples. In this way, AOX also provides a degree of signaling homeostasis to the organelle. Evidence suggests that AOX function in metabolic and signaling homeostasis is particularly important during stress. These include abiotic stresses such as low temperature, drought, and nutrient deficiency, as well as biotic stresses such as bacterial infection. This review provides an introduction to the genetic and biochemical control of AOX respiration, as well as providing generalized examples of how AOX activity can provide metabolic and signaling homeostasis. -

The Role of Hydroxamic Acids in Biochemical Processes
Biochemistry THE ROLE OF HYDROXAMIC ACIDS IN BIOCHEMICAL PROCESSES AHMED E. FAZARY* MOHAMED M. KHALIL** ALY FAHMY* TANTAWY A. TANTAWY* SUMMARY: Hydroxamic acids, a group of naturally occurring and synthetic weak organic acids of general formula RC(=O)N(R')OH, are widespread in the tissues of plants, in metabolites of bacteria and fungi, including complex compounds. Hydroxamic acids and their derivatives fulfill a variety of important roles in biology and medicine; here we provide a comprehensive brief review of the most basic medicinal chemistry and pharmacology of hydroxamate molecules. Key Words: Hydroxamic acids, biological activities, biochemical processes. INTRODUCTION INHIBITION EFFECT AND ANTICANCER ACTIVITY Since their discovery by Wahlroos and Virtanen (1) in OF HYDROXAMIC ACIDS 1959, and over the past decades, the chemistry and bio- The design and synthesis of ligands for biomedical chemistry of hydroxamic acids and their derivatives have applications in fields such as anticancer applications has attracted considerable attention, due to their pharmaco- become of great importance. One of these important lig- logical, toxicological and pathological properties. Hydrox- ands is hydroxamate molecules. Hydroxamic acids have amic acids generally have low toxicities and have a wide been found to react with both proteins and nucleic acids spectrum of activities in all types of biological systems, as (2). The reactivity of hydroxamic acids towards sulfhydryl such they act variously as growth factors, food additives, groups of proteins has been suggested to be the reason tumor inhibitors, antimicrobial agents, antituberculous, for their inhibitory effect on various enzymes. The pro- antileukemic agents, key pharmacophore in many impor- tease papain, for instance, with a single free cysteine tant chemotherapeutic agents, pigments and cell-division residue located at the active site was irreversibly inhibited factors. -

A Review on the Metal Complex of Nickel (II) Salicylhydroxamic Acid and Its Aniline Adduct
Adegoke AV, et al., J Transl Sci Res 2019, 2: 006 DOI: 10.24966/TSR-6899/100006 HSOA Journal of Translational Science and Research Review Article Introduction A Review on the Metal Complex Metal complexes are formed in biological systems, particularly of Nickel (II) Salicylhydroxamic between ligands and metal ions in dynamic equilibrium, with the free metal ion in a more or less aqueous environment [1]. All biologically Acid and its Aniline Adduct important metal ions can form complexes and the number of different chemical species which can be coordinated with these metal ions is very large. During the past few decades, a lot of scientist research 1 1 2 3 Adegoke AV *, Aliyu DH , Akefe IO and Nyan SE groups operated through specialization in the direction of drug dis- 1Department of Chemistry, Faculty of Science, University of Abuja, Nigeria covery, by studying the simplest species that use metal ions and re- searching them as whole compound; for example, they suggested the 2 Department of Physiology, Biochemistry and Pharmacology, Faculty of addition of metal ion to antibiotics to facilitate their spread through- Veterinary Medicine, University of Jos, Nigeria out the body [2]. The development of drug resistance as well as the 3Department of Chemistry, Faculty of Science, Kaduna State University, appearance of undesirable side effects of certain antibiotics has led to Nigeria the search of new antimicrobial agents with the goal to discover new chemical structures which overcome the above disadvantages. As a ligand with potential oxygen and nitrogen donors, hydroxamic acids are interesting and have gained special attention not only because of Abstract the structural chemistry of their coordination modes, but also because Metal complexes are fundamentally known to be engendered in of their importance in medical chemistry [3]. -

Wo 2008/127291 A2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (43) International Publication Date PCT (10) International Publication Number 23 October 2008 (23.10.2008) WO 2008/127291 A2 (51) International Patent Classification: Jeffrey, J. [US/US]; 106 Glenview Drive, Los Alamos, GOlN 33/53 (2006.01) GOlN 33/68 (2006.01) NM 87544 (US). HARRIS, Michael, N. [US/US]; 295 GOlN 21/76 (2006.01) GOlN 23/223 (2006.01) Kilby Avenue, Los Alamos, NM 87544 (US). BURRELL, Anthony, K. [NZ/US]; 2431 Canyon Glen, Los Alamos, (21) International Application Number: NM 87544 (US). PCT/US2007/021888 (74) Agents: COTTRELL, Bruce, H. et al.; Los Alamos (22) International Filing Date: 10 October 2007 (10.10.2007) National Laboratory, LGTP, MS A187, Los Alamos, NM 87545 (US). (25) Filing Language: English (81) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of national protection available): AE, AG, AL, AM, AT,AU, AZ, BA, BB, BG, BH, BR, BW, BY,BZ, CA, CH, (30) Priority Data: CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, EG, 60/850,594 10 October 2006 (10.10.2006) US ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, KZ, LA, LC, LK, (71) Applicants (for all designated States except US): LOS LR, LS, LT, LU, LY,MA, MD, ME, MG, MK, MN, MW, ALAMOS NATIONAL SECURITY,LLC [US/US]; Los MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PG, PH, PL, Alamos National Laboratory, Lc/ip, Ms A187, Los Alamos, PT, RO, RS, RU, SC, SD, SE, SG, SK, SL, SM, SV, SY, NM 87545 (US). -

A Review on Hydroxamic Acids
INTERNATIONAL JOURNAL OF BIOLOGY AND BIOMEDICAL ENGINEERING DOI: 10.46300/91011.2020.14.12 Volume 14, 2020 A review on Hydroxamic Acids: Widespectrum Chemotherapeutic Agents Zainab Syed1, 2, Kumar Sonu1, Aman Dongre1, 2 , Gopesh Sharma2, Monika Sogani1* 1Schoolof Civil and Chemical Engineering, Manipal University Jaipur, Rajasthan 300307, India 2Department of Biosciences, Manipal University Jaipur, Rajasthan 300307, India *Author for correspondence: Tel: +919414207450 Received: January 28, 2020. Revised: March 10, 2020. 2nd Revised: May 25, 2020. Accepted: June 22, 2020. Published: July 20, 2020. deformilases or enzyme inhibitors [1]. Various substituted Abstract: Recent developments in drug discovery analogues of hydroxamic acid are reported to possess anti- have highlighted the ability of hydroxamic acids to form inflammatory, anti-tubercular, anti-leukemic, anti-microbial complexes with various metal ions, in particular iron, and tumor inhibiting properties. They play a vital role in the zinc, magnesium and calcium, and this imparts them manufacturing of various biologically active drugs as anti- with a number of unique biological and pharmacological depressants, anti-tumor agents, anti-HIV agents and anti- properties. This review provides information on the malarial agents and are also used in industries as most significant developments of the hydroxamate insecticides, antioxidants, as inhibitors of corrosion and for compounds in the medicinal area with a focus on the extraction of toxic elements [2]. Suberoylanilide hydroxamic acid (SAHA) and its derivatives with antibacterial, antifungal, antiviral, II. BIOLOGICAL AND MEDICINAL SIGNIFICANCE OF antimalarial, antitubercular and anticancer effect and HYDROXAMIC ACIDS their possible molecular mechanisms. One of the latest Although they have been known from long ago to be favorable developments has been on developing SAHA biologically active, their varied biological features are still and its hybrids as potent anti-tuberculosis drugs. -
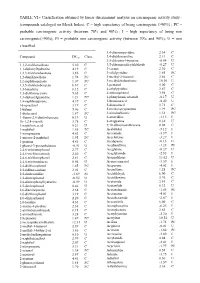
TABLE VI.- Classification Obtained by Linear Discriminant Analysis on Carcinogenic Activity Study : (Compounds Cataloged on Merck Index)
TABLE VI.- Classification obtained by linear discriminant analysis on carcinogenic activity study : (compounds cataloged on Merck Index). C = high expectancy of being carcinogenic (>90%) ; PC = probable carcinogenic activity (between 70% and 90%) ; I = high expectancy of being non carcinogenic(>90%); PI = probable non carcinogenic activity (between 70% and 90%); U = non classified. 3,4-diaminopyridine 2.34 C Compound DFcarc Class. 3,4-dichloroaniline 2.13 C 3,5-dibromo-l-tyrosine -0.54 U 1,1,2-trichloroethane 5.30 C 3,5-dibromosalicylaldehyde -0.27 U 1,1-diphenylhydrazine 4.19 C 3-carene 2.30 C 1,2,3-trichlorobenzene 3.86 C 3-ethylpyridine 1.85 PC 1,2-dianilinoethane 1.74 PC 3-methyl-2-butanol 3.66 C 1,2-naphthoquinone 1.07 PC 3-methylcholanthrene 10.30 U 1,3,5-trichlorobenzene 6.67 C 3-pentanol 5.00 C 1,3-butadiene 8.12 C 4-ethylpyridine 2.87 C 1,3-dichloroacetone 5.65 C 4-nitrosophenol 3.98 C 1,3-diphenylguanidine 1.21 PC 4-phenylsemicarbazide -0.17 U 1,4-naphthoquinone 4.37 C 5-bromouracil -0.42 U 16-epiestriol 3.19 C 5-diazouracil 3.71 C 1-butene 5.06 C 5-methoxytryptamine 1.99 PC 1-dodecanol 1.87 PC 5-nitrobarbituric 1.51 PC 1-fluoro-2,4-dinitrobenzene 0.19 U 6-azauridine -3.15 I 1h-1,2,4-triazole 3.75 C 8-azaguanine 0.30 U 1-naphthoic,acid 0.21 U 9,10-dibromoanthracene 8.02 C 1-naphthol 1.65 PC Acebutolol -5.12 I 1-nitropropane 4.63 C Acecainide -3.39 I 1-nitroso-2-naphthol 1.95 PC Aceclofenac -3.27 I 1-pentene 4.41 C Acedapsone -0.13 U 1-phenyl-3-pyrazolidinone -0.91 U Acediasulfone -1.23 PI 2,4,6-tribromophenol 2.77 C Aceglatone -

Localization and Functional Characterization of the Alternative Oxidase in Naegleria
bioRxiv preprint doi: https://doi.org/10.1101/2020.09.26.314807; this version posted September 26, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Localization and Functional Characterization of the Alternative 2 Oxidase in Naegleria 3 4 Running title: Naegleria gruberi’s AOX 5 6 Diego Cantonia,1, Ashley Osbornea,1, Najwa Taibb,c, Gary Thompsond, Eleanna Kazanaa, Elizabeth 7 Edriche, Ian R. Brownf, Simonetta Gribaldob, Campbell W Gourlaye, and Anastasios D. Tsaousisa 8 9 a Laboratory of Molecular & Evolutionary Parasitology, RAPID group, School of Biosciences, 10 University of Kent, Canterbury, CT2 7NJ, UK 11 b Unit Evolutionary Biology of the Microbial Cell, Department of Microbiology, Institut Pasteur, 12 UMR CNRS 2001, Paris, France. 13 c 14 Hub Bioinformatics and Biostatistics, Department of Computational Biology, Institut Pasteur, USR 15 3756 CNRS, Paris, France. 16 17 d NMR facility, School of Biosciences, University of Kent, Canterbury CT2 7NJ, UK. 18 19 e Kent Fungal Group, RAPID group, School of Biosciences, University of Kent, Canterbury CT2 20 7NJ, UK. 21 22 f Bioimaging Facility, School of Biosciences, University of Kent, Canterbury, CT2 7NJ, UK 23 24 25 1Equal contribution 26 27 * Correspondence: 28 Dr. Anastasios D. Tsaousis, RAPID group, School of Biosciences, University of Kent, Canterbury, 29 Kent, UK, Telephone number: +44 1227 827007; email: [email protected] / 30 [email protected] 31 32 33 Keywords 34 Naegleria, Respiration; mitochondria; confocal microscopy; evolution; adaptation 35 36 37 38 39 40 41 42 43 44 45 46 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.09.26.314807; this version posted September 26, 2020. -

Neurogenesis Via Modulation of the Muscarinic Receptors
(19) & (11) EP 2 275 096 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 19.01.2011 Bulletin 2011/03 A61K 31/00 (2006.01) A61K 31/66 (2006.01) A61K 31/325 (2006.01) A61K 31/473 (2006.01) (2006.01) (2006.01) (21) Application number: 10009737.7 A61K 31/445 A61P 25/00 (22) Date of filing: 25.08.2006 (84) Designated Contracting States: • Pires, Jammieson C. AT BE BG CH CY CZ DE DK EE ES FI FR GB GR San Diego HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI California 92139 (US) SK TR • Morse, Andrew San Diego (30) Priority: 26.08.2005 US 711846 P California 92128 (DE) 14.10.2005 US 727127 P • Gitnick, Dana 17.11.2005 US 738133 P San Marcos 02.06.2006 US 803826 P California 92078 (US) • Treuner, Kai (62) Document number(s) of the earlier application(s) in 5204 San Diego accordance with Art. 76 EPC: California 92122 (US) 06813787.6 / 1 928 437 • Broadhead, Alex San Diego (71) Applicant: Braincells, Inc. California 92117 (US) San Diego, CA 92121 (US) (74) Representative: Roques, Sarah Elizabeth (72) Inventors: J.A. Kemp & Co. • Barlow, Carrolee 14 South Square Del Mar Gray’s Inn California 92129 (US) London WC1R 5JJ (GB) • Carter, Todd A. San Diego Remarks: California 92129 (US) This application was filed on 16-09-2010 as a • Lorrain, Kym I. divisional application to the application mentioned San Diego under INID code 62. California 92124 (US) (54) Neurogenesis via modulation of the muscarinic receptors (57) The instant disclosure describes methods for as via inhibition of acetylcholine esterase (AChE) activity, treating diseases and conditions of the central and pe- alone or in combination with another neurogenic agent ripheral nervous system by stimulating or increasing neu- to stimulate or activate the formation of new nerve cells. -

Modern Fungicides and Antifungal Compounds VIII Persistent Identifier: Urn:Nbn:De:0294-Sp-2017-Reinh-4 Arc I Spectrum Phytomedizin
Persistent Identifier: urn:nbn:de:0294-sp-2017-Reinh-4 ore I Spectrum Phytomedizin H.B. Deising, B. Fraaije, A. Mehl, E.C. Oerke, H. Sierotzld, G. Stammler Modern Fungicides and Antifungal Compounds VIII Persistent Identifier: urn:nbn:de:0294-sp-2017-Reinh-4 arc I Spectrum Phytomedizin Proceedings of the 18th International Reinhardsbrunn Symposium on Modern Fungicides and Antifungal Cam pounds 2016 'I11e iii-annual Relnhardsbnlnn Symposium hal a longstanding b'adition and is the leading international meeting focusing on fungicide science today. Participants from twenty-six different counb'ies around the globe presented more than eighty outstanding sclentlftc papers. 'I11ese papers covered topics on different modes of fungicide resistance, sensitivity monitoring, resistance management strategies, and new applications and technologies. 'I11ese exciting scientific topics are captured in this IJIh volume of the Modem FUngicides and AntU'ungal Compounds series. 1be outstanding written contributions of all pres! .teis at the symposium demonsb'ate not only the excellence of expe rienced --.but also the brilliance of younger scientists in the Increasingly important field of plant protection. Persistent Identifier: urn:nbn:de:0294-sp-2017-Reinh-4 DPG Spectrum Phytomedizin Deising H.B.; Fraaije B.; Mehl A.; Oerke E.C.; Sierotzki H.; Stammler G. Modern Fungicides and Antifungal Compounds VIII Proceedings of the 18th International Reinhardsbrunn Symposium April 24-28, 2016, Friedrichroda, Germany Publisher Persistent Identifier: urn:nbn:de:0294-sp-2017-Reinh-4 Bibliografische Information der Deutschen Bibliothek Die Deutsche Bibliothek verzeichnet diese Publikation in der Deutschen Nationalbibliografie Detaillierte bibliografische Daten sind im Internet über http://dnb.ddb.de abrufbar. ISBN: 978-3-941261-15-0 Das Werk einschließlich aller Teile ist urheberrechtlich geschützt. -

Salicylhydroxamic Acid Inhibits the Growth of Candida Albicans
World Academy of Science, Engineering and Technology International Journal of Bioengineering and Life Sciences Vol:5, No:4, 2011 Salicylhydroxamic Acid Inhibits the Growth of Candida albicans Shu-Ying Marissa Pang, Stephen Tristram and Simon Brown chain enzyme that accepts electrons directly from ubiquinol Abstract—Candida spp. are common and aggressive pathogens. (UQH2) and reduces O2 to water. Because of the growing resistance of Candida spp. to current Alternative oxidase activity is inhibited by compounds such antifungals, novel targets, found in Candida spp. but not in humans as salicylhydroxamic acid (SHAM) [4], disulfiram and n-alkyl or other flora, have to be identified. The alternative oxidase (AOX) gallates [5, 6], none of which inhibit the activity of is one such possibility. This enzyme is insensitive to cyanide, but is sensitive to compounds such as salicylhydroxamic acid (SHAM), cytochrome oxidase. Conversely, cyanide inhibits disulfiram and n-alkyl gallates. The growth Candida albicans was cytochrome oxidase, but does not inhibit AOX. These two inhibited by SHAM (Ki = 9-15 mM) and cyanide (Ki = 2-4 mM), inhibitors can be used to distinguish between O2 uptake albeit to differing extents. The rate of O2 uptake was inhibited by catalysed by AOX and cytochrome oxidase. The disruption of less than 10% by 25 mM SHAM and by about 90% by 250 μM electron transfer by cyanide and SHAM would affect ATP KCN. Although SHAM substantially inhibited the growth of C. synthesis and the activity of metabolic pathways, either of albicans, it is unlikely that the inhibition of AOX was the cause. which would inhibit growth. -

Table of Drugs and Chemicals (FY12)
ICD-9-CM Table of Drugs and Chemicals (FY12) Table of Drugs and Chemicals INDEX TO POISONING AND EXTERNAL CAUSES OF ADVERSE EFFECTS OF DRUGS AND OTHER CHEMICAL SUBSTANCES This table contains a classification of drugs and other chemical substances to identify poisoning states and external causes of adverse effects. Each of the listed substances in the table is assigned a code according to the poisoning classification (960-989). These codes are used when there is a statement of poisoning, overdose, wrong substance given or taken, or intoxication. The table also contains a listing of external causes of adverse effects. An adverse effect is a pathologic manifestation due to ingestion or exposure to drugs or other chemical substances (e.g., dermatitis, hypersensitivity reaction, aspirin gastritis). The adverse effect is to be identified by the appropriate code found in Section 1, Index to Diseases and Injuries. An external cause code can then be used to identify the circumstances involved. The table headings pertaining to external causes are defined below: Accidental poisoning (E850-E869)accidental overdose of drug, wrong substance given or taken, drug taken inadvertently, accidents in the usage of drugs and biologicals in medical and surgical procedures, and to show external causes of poisonings classifiable to 980-989. Therapeutic use (E930-E949)a correct substance properly administered in therapeutic or prophylactic dosage as the external cause of adverse effects. Suicide attempt (E950-E952)instances in which self-inflicted injuries or poisonings are involved. Assault (E961-E962)injury or poisoning inflicted by another person with the intent to injure or kill. -

List/Code of Hazardous Materials
Hazardous Materials Table This Table is a compilation of lists of hazardous materials from the following sources: 1. Environmental Protection Agency (EPA), Title 40 Code of Federal Regulations, Hazardous Waste Regulations 2. Department of Transportation (DOT), Title 49 Code of Federal Regulations, Transportation of Hazardous Materials 3. Califonia Medical Waste Management Act (MWMA) 4. Department of Labor, Occupational Safety & Health Administration, Title 29 Code of Federal Regulations, Subpart Z, Toxic and Hazardous Substances 5. Environmental Protection Agency, Superfund Amendment & Reauthorization Act (SARA)/Title III, Extremely Hazardous Substances 6. American Conference of Governmental Industrial Hygienists, Identification and classification of carcinogens. (1986) Key to Hazard Codes C Corrosive CL Combustible Liquid E Explosive FG Flammable Gas FL Flammable Liquid FS Flammable Solid I Irritating Material NFG Nonflammable Gas OA Otherwise Regulated Material Class A OB Otherwise Regulated Material Class B OC Otherwise Regulated Material Class C OD Otherwise Regulated Material Class D OE Otherwise Regulated Material Class E OG Organic Peroxide OX Oxidizer P Poison R Reactive * Nonhazardous Waste Note: Materials without a hazard code have not been classified and may be hazardous. 1 Hazardous Materials Table N.O.S. Descriptions Acetorphine Acetoxytriphenylstannane Flammable liquid, n.o.s. (FL) Acetyl Bromide (C) Combustible liquid, n.o.s. (CL) Acetyl Chloride (FL, P) Hazardous Waste Liquid, n.o.s. (OE) Acetyl Iodide (C) Acid liquid, n.o.s. (C) Acetylacetone (FL) Flammable liquid, corrosive, n.o.s. (FL, C) 2-Acetylaminofluorene (P) Hazardous Waste Solid, n.o.s. (OE) Acetylbromazine Formaldehyde solution (P, OA) Acetyldihydrocodeine Corrosive solids, n.o.s. (C) Acetylene (FG) Flammable liquid, poisonous, n.o.s.