Cain Help Index
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Tripwire Connect 3.0.1 Installation Guide
Appendix I. Third-Party License Agreements The Apache Software License, Version 2.0 This product includes software licensed under the Apache License, Version 2.0 (the "License"); you may not use this software except in compliance with the License. You may obtain a copy of the License at http://www.apache.org/licenses/LICENSE-2.0 Unless required by applicable law or agreed to in writing, software distributed under the License is distributed on an "AS IS" BASIS, WITHOUT WARRANTIES OR CONDITIONS OF ANY KIND, either express or implied. See the License for the specific language governing permissions and limitations under the License. Apache Batik Copyright 1999-2007 The Apache Software Foundation This product includes software developed at The Apache Software Foundation (http://www.apache.org/). This software contains code from the World Wide Web Consortium (W3C) for the Document Object Model API (DOM API) and SVG Document Type Definition (DTD). This software contains code from the International Organisation for Standardization for the definition of character entities used in the software's documentation. This product includes images from the Tango Desktop Project (http://tango.freedesktop.org/). This product includes images from the Pasodoble Icon Theme (http://www.jesusda.com/projects/pasodoble). AntiSamy Copyright (c) 2007-2012, Arshan Dabirsiaghi, Jason Li All rights reserved. Redistribution and use in source and binary forms, with or without modification, are permitted provided that the following conditions are met: - Redistributions of source code must retain the above copyright notice, this list of conditions and the following disclaimer. - Redistributions in binary form must reproduce the above copyright notice, this list of conditions and the following disclaimer in the documentation and/or other materials provided with the distribution. -

DCP-O-Matic Users' Manual
DCP-o-matic users’ manual Carl Hetherington DCP-o-matic users’ manual ii Contents 1 Introduction 1 1.1 What is DCP-o-matic?.............................................1 1.2 Licence.....................................................1 1.3 Acknowledgements...............................................1 1.4 This manual...................................................1 2 Installation 2 2.1 Windows....................................................2 2.2 Mac OS X....................................................2 2.3 Debian, Ubuntu or Mint Linux........................................2 2.4 Fedora, Centos and Mageia Linux.......................................2 2.5 Arch Linux...................................................3 2.6 Other Linux distributions...........................................3 2.7 ‘Simple’ and ‘Full’ modes...........................................4 3 Creating a DCP from a video 5 3.1 Creating a new film..............................................5 3.2 Adding content.................................................6 3.3 Making the DCP................................................7 4 Creating a DCP from a still image9 5 Manipulating existing DCPs 12 5.1 Importing a DCP into DCP-o-matic..................................... 12 5.2 Decrypting encrypted DCPs.......................................... 12 5.3 Making a DCP from a DCP.......................................... 12 5.3.1 Re-use of existing data......................................... 13 5.3.2 Making overlay files......................................... -

Curriculum Vitae ( PDF )
Simon Pascal Klein Last updated: Tuesday, 6 May 2014 ! http://klepas.org Nationality: German (Australian permanent resident) " [email protected] Current residence: Canberra, Australia # +61 420 9797 38 ABN: 94 483 395 962 I’m Pascal — a front-end designer & writer, with a penchant for accessibility and typography. tl;dr? Both in form & content, I build beautiful and usable interfaces. Skils overview writing, research, Rooted in the open source & web community. policy advice accessibility, documentation, teaching, open source, gender, … front-end web Semantic, standards-based, accessible, and responsive web development. development & HTML, css, SVG; WCAG2; Drupal, Jekyll, WordPress, …; terminal, git, … accessibility design & media Content & media curation; design direction; podcasting, pedantic copy editing. curation the , Photoshop; Inkscape, Illustrator; audio editing, Markdown, … ! gimp ! Experience 2013 Senior Policy Advisor & web specialist — act Government Reporting to the cto, I worked for the Chief Minister and Treasury Directorate in a policy and research role as a web technologies and accessibility specialist. Work also included web development on internal prototyping projects that un- derpinned Open Government and Open Data mandates. I also launched the frst Australian government GitHub account, for the act. 2011 Managing Editor, podcast host & producer — SitePoint Lead editor & manager for SitePoint’s then new DesignFestival.com. I launched the site’s podcast, producing 15 episodes (hiting 30,000 downloads before leav- ing this position). Additionaly I authored original articles for SitePoint. I also freelanced during this time. !1 2007–09 Concept Designer — Looking Glass Solutions Graphic & interface designer, embedded within the development team. lgs was a former web development studio that serviced primarily the Australian Government and Engineers Australia. -
Soatopicindex - QVIZ
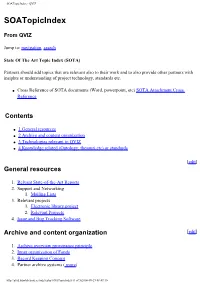
SOATopicIndex - QVIZ SOATopicIndex From QVIZ Jump to: navigation, search State Of The Art Topic Index (SOTA) Partners should add topics that are relevant also to their work and to also provide other partners with insights or understanding of project technology, standards etc. ● Cross Reference of SOTA documents (Word, powerpoint, etc) SOTA Attachment Cross Reference Contents ● 1 General resources ● 2 Archive and content organization ● 3 Technologies relevant to QVIZ ● 4 Knowledge related (Ontology, thesauri,etc) or standards [edit] General resources 1. Relvant State-of-the-Art Reports 2. Support and Networking 1. Mailing Lists 3. Relevant projects 1. Electronic library project 2. Relevant Projects 4. Issue and Bug Tracking Software Archive and content organization [edit] 1. Archive overview provenance principle 2. Inner organization of Fonds 3. Record Keeping Concept 4. Partner archive systems ( more) http://qviz.humlab.umu.se/index.php/SOATopicIndex (1 of 5)2006-09-29 08:49:10 SOATopicIndex - QVIZ 1. NAE System Description 2. SVAR and National Archives System Description 3. Vision of Britain System Description 4. Comparison of admin unit issues across partners systems 5. Archive features - QVIZ 1. Trackback 6. Archive Standards Technologies relevant to QVIZ [edit] 1. Image annotation to support user generated Thematic maps 2. Web Tools: Screen Capture 3. Prominent digital repositories Technologies and Digital Archive Technologies 1. See also Digital Object Metadata 4. Semantic repositories and other basis repository techologies (also semantic digital or semantic e-Libraries, etc) 5. Semantic web services (SWS) and Service oriented architecture (SOA) 6. Access Stategies 7. Workflow Technologies (includes BEPL,etc related tools) 8. Relevant social software (mainly to point out relevant features) 1. -

Engaging Developers in Open Source Software Projects: Harnessing Social
Iowa State University Capstones, Theses and Graduate Theses and Dissertations Dissertations 2015 Engaging developers in open source software projects: harnessing social and technical data mining to improve software development Patrick Eric Carlson Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/etd Part of the Computer Engineering Commons, Computer Sciences Commons, and the Library and Information Science Commons Recommended Citation Carlson, Patrick Eric, "Engaging developers in open source software projects: harnessing social and technical data mining to improve software development" (2015). Graduate Theses and Dissertations. 14663. https://lib.dr.iastate.edu/etd/14663 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Engaging developers in open source software projects: Harnessing social and technical data mining to improve software development by Patrick Eric Carlson A dissertation submitted to the graduate faculty in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Major: Human-Computer Interaction Program of Study Committee: Judy M. Vance, Major Professor Tien Nguyen James Oliver Jon Kelly Stephen Gilbert Iowa State University Ames, Iowa 2015 Copyright c Patrick Eric Carlson, 2015. All rights reserved. ii DEDICATION This is dedicated to my parents who have always supported me. iii TABLE OF CONTENTS Page LIST OF TABLES . vi LIST OF FIGURES . vii ACKNOWLEDGEMENTS . ix ABSTRACT . x CHAPTER 1. -

Vsim Installation and Release Notes Release 10.1.0-R2780
VSim Installation and Release Notes Release 10.1.0-r2780 Tech-X Corporation Mar 12, 2020 2 CONTENTS 1 Overview 1 2 Installation Details 3 2.1 What is Installed with VSim?......................................3 2.2 VSim System Requirements.......................................4 2.3 VSim Installation Instructions......................................5 2.4 VSim Documentation.......................................... 10 2.5 VSim Release Notes........................................... 12 2.6 Software Licensing............................................ 30 3 Trademarks and licensing 45 i ii CHAPTER ONE OVERVIEW This guide shows how to customize VSim [VSi], the arbitrary dimensional, electromagnetics and plasma simulation code, to add macros and analyzers. VSim [VSi] is an arbitrary dimensional, electromagnetics and plasma simulation code consisting of two major com- ponents: • VSimComposer, the graphical user interface. • Vorpal [NC04], the VSim Computational Engine. VSim also includes many more items such as Python, MPI, data analyzers, and a set of input simplifying macros. 1 VSim Installation and Release Notes, Release 10.1.0-r2780 2 Chapter 1. Overview CHAPTER TWO INSTALLATION DETAILS 2.1 What is Installed with VSim? Upon completing the installation process (described in VSim Installation Instructions), VSimComposer, the VSim Computation Engine, Python, and MPI will be installed on your computer. These are described in detail below. 2.1.1 VSimComposer VSimComposer is a graphical user interface for • Creating and editing VSim input files • Executing VSim • Analyzing VSim generated data • Visualizing VSim generated data • Viewing the documentation. The VSimComposer editor and validator have built-in functions and graphical components that help you to create input files. Example input files, ranging in complexity from beginning to advanced, are included with VSimComposer. New VSim users can use these examples as templates. -

Exploring Usability Guidelines for Rich Internet Applications
Department of Informatics Exploring Usability Guidelines for Rich Internet Applications Master thesis, 10 credits, Department of Informatics Presented: June 2007 Authors: Lukas Gwardak Linus Påhlstorp Supervisor: Konrad Tollmar LUND UNIVERSITY Informatics Exploring Usability Guidelines for Rich Internet Applications Master thesis, presented June, 2007 Supervisor: Konrad Tollmar Size: 80 pages Abstract: Usability guidelines are commonly considered a useful tool for developers to enhance the usability of interactive systems. They represent distilled knowledge from many disciplines related to usability and provide developers with solutions and best practices to achieve usability goals. However, the newly developing field of highly interactive web applications (Rich Internet Applications) still lacks appropriate usability guidelines. This work takes desktop usability guidelines and web usability guidelines as a basis to create an outline of Rich Internet Application usability guidelines. Three professional developers are being interviewed in order to get an insight into their work with guidelines and get their ideas of how possible Rich Internet Application guidelines should be structured. Keywords: Usability, accessibility, guidelines, user interface, web design, Rich Internet Applications, Web 2.0 Contents 2 Contents Contents...................................................................................................................... 2 Index of Figures......................................................................................................... -
Free and Open Source Software
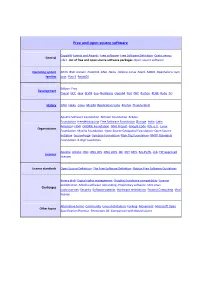
Free and open source software Copyleft ·Events and Awards ·Free software ·Free Software Definition ·Gratis versus General Libre ·List of free and open source software packages ·Open-source software Operating system AROS ·BSD ·Darwin ·FreeDOS ·GNU ·Haiku ·Inferno ·Linux ·Mach ·MINIX ·OpenSolaris ·Sym families bian ·Plan 9 ·ReactOS Eclipse ·Free Development Pascal ·GCC ·Java ·LLVM ·Lua ·NetBeans ·Open64 ·Perl ·PHP ·Python ·ROSE ·Ruby ·Tcl History GNU ·Haiku ·Linux ·Mozilla (Application Suite ·Firefox ·Thunderbird ) Apache Software Foundation ·Blender Foundation ·Eclipse Foundation ·freedesktop.org ·Free Software Foundation (Europe ·India ·Latin America ) ·FSMI ·GNOME Foundation ·GNU Project ·Google Code ·KDE e.V. ·Linux Organizations Foundation ·Mozilla Foundation ·Open Source Geospatial Foundation ·Open Source Initiative ·SourceForge ·Symbian Foundation ·Xiph.Org Foundation ·XMPP Standards Foundation ·X.Org Foundation Apache ·Artistic ·BSD ·GNU GPL ·GNU LGPL ·ISC ·MIT ·MPL ·Ms-PL/RL ·zlib ·FSF approved Licences licenses License standards Open Source Definition ·The Free Software Definition ·Debian Free Software Guidelines Binary blob ·Digital rights management ·Graphics hardware compatibility ·License proliferation ·Mozilla software rebranding ·Proprietary software ·SCO-Linux Challenges controversies ·Security ·Software patents ·Hardware restrictions ·Trusted Computing ·Viral license Alternative terms ·Community ·Linux distribution ·Forking ·Movement ·Microsoft Open Other topics Specification Promise ·Revolution OS ·Comparison with closed -

"Accept" Or "Agree" B
CAREFULLY READ THIS COLLECTION OF INFORMATION AND LICENSE AGREEMENTS. BY CLICKING THE "ACCEPT" OR "AGREE" BUTTON, OR OTHERWISE ACCESSING, DOWNLOADING, INSTALLING OR USING THE SOFTWARE, YOU AGREE ON BEHALF OF LICENSEE TO BE BOUND BY THIS INFORMATION AND LICENSE AGREEMENTS (TO THE EXTENT APPLICABLE TO THE SPECIFIC SOFTWARE YOU OBTAIN AND THE SPECIFIC MANNER IN WHICH YOU USE SUCH SOFTWARE). IF LICENSEE DOES NOT AGREE TO ALL OF THE INFORMATION AND LICENSE AGREEMENTS BELOW, DO NOT CLICK THE "ACCEPT" OR "AGREE" BUTTON OR ACCESS, DOWNLOAD, INSTALL OR USE THE SOFTWARE; AND IF LICENSEE HAS ALREADY OBTAINED THE SOFTWARE FROM AN AUTHORIZED SOURCE, PROMPTLY RETURN IT FOR A REFUND. Part One: Overview. The following information applies to certain items of third-party technology that are included along with certain Xilinx software tools. The Xilinx Embedded Development Kit (EDK) is a suite of software and other technology that enables Licensee to design a complete embedded processor system for use in a Xilinx Device. EDK includes, among other components, (a) the Xilinx Platform Studio (XPS), which is the development environment, or GUI, used for designing the hardware portion of an embedded processor system; and (b) the Xilinx Software Development Kit (SDK), which is an integrated development environment, complementary to XPS, that is used to create and verify C/C++ embedded software applications. SDK is also made available separately from EDK. Licensee's use of the GNU compilers (including associated libraries and utilities) that are supplied with SDK may cause Licensee's software application (or board- support package) to be governed by certain third-party "open source" license agreements, as further described below. -
TE Console 8.7.2 - Use of Third Party Libraries
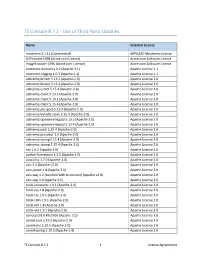
TE Console 8.7.2 - Use of Third Party Libraries Name Selected License mindterm 4.1.12 (Commercial) APPGATE-Mindterm-License GifEncoder 1998 (Acme.com License) Acme.com Software License ImageEncoder 1996 (Acme.com License) Acme.com Software License commons-discovery 0.2 (Apache 1.1) Apache License 1.1 commons-logging 1.0.3 (Apache 1.1) Apache License 1.1 activemq-broker 5.13.2 (Apache-2.0) Apache License 2.0 activemq-broker 5.15.4 (Apache-2.0) Apache License 2.0 activemq-camel 5.15.4 (Apache-2.0) Apache License 2.0 activemq-client 5.13.2 (Apache-2.0) Apache License 2.0 activemq-client 5.14.2 (Apache-2.0) Apache License 2.0 activemq-client 5.15.4 (Apache-2.0) Apache License 2.0 activemq-jms-pool 5.15.4 (Apache-2.0) Apache License 2.0 activemq-kahadb-store 5.15.4 (Apache-2.0) Apache License 2.0 activemq-openwire-legacy 5.13.2 (Apache-2.0) Apache License 2.0 activemq-openwire-legacy 5.15.4 (Apache-2.0) Apache License 2.0 activemq-pool 5.15.4 (Apache-2.0) Apache License 2.0 activemq-protobuf 1.1 (Apache-2.0) Apache License 2.0 activemq-spring 5.15.4 (Apache-2.0) Apache License 2.0 activemq-stomp 5.15.4 (Apache-2.0) Apache License 2.0 ant 1.6.3 (Apache 2.0) Apache License 2.0 avalon-framework 4.2.0 (Apache 2.0) Apache License 2.0 awaitility 1.7.0 (Apache-2.0) Apache License 2.0 axis 1.4 (Apache v2.0) Apache License 2.0 axis-jaxrpc 1.4 (Apache 2.0) Apache License 2.0 axis-saaj 1.2 [bundled with te-console] (Apache v2.0) Apache License 2.0 axis-saaj 1.4 (Apache 2.0) Apache License 2.0 batik-constants 1.9.1 (Apache-2.0) Apache License 2.0 batik-css -

Java SE Licensing Information User Manual
Licensing Information User Manual Oracle Java SE and Oracle Java SE Embedded Products Last updated: 2021/07/20 Contents Title and Copyright Information Introduction Licensing Information Description of Product Editions and Permitted Features Prerequisite Products Entitled Products and Restricted Use Licenses Third Party Notices and/or Licenses Title and Copyright Information Introduction This Licensing Information document is a part of the product or program documentation under the terms of your Oracle license agreement and is intended to help you understand the program editions, entitlements, restrictions, prerequisites, special license rights, and/or separately licensed third party technology terms associated with the Oracle software program(s) covered by this document (the “Program(s)”). Entitled or restricted use products or components identified in this document that are not provided with the particular Program may be obtained from the Oracle Software Delivery Cloud website (https://edelivery.oracle.com) or from media Oracle may provide. If you have a question about your license rights and obligations, please contact your Oracle sales representative, review the information provided in Oracle’s Software Investment Guide (http://www.oracle.com/us/corporate/pricing/software-investment-guide/index.html), and/or contact the applicable Oracle License Management Services representative listed on http://www.oracle.com/us/corporate/license-management- services/index.html Licensing Information Description of Product Editions and Permitted Features The Oracle Java SE and Oracle Java SE Embedded products listed below include multiple features. In order to gain access to all of the features applicable to the product you have licensed, you may need to download more than one installation package. -

DXR Documentation Release 2.0
DXR Documentation Release 2.0 various Aug 27, 2018 Contents 1 Contents 3 1.1 Welcome To The DXR Community...................................3 1.2 Getting Started..............................................4 1.3 Configuration...............................................6 1.4 Deployment............................................... 10 1.5 Use.................................................... 15 1.6 Development............................................... 18 1.7 Appendix A: Indexing Firefox...................................... 33 2 Back Matter 35 2.1 Glossary................................................. 35 2.2 Icon Credits............................................... 35 i ii DXR Documentation, Release 2.0 DXR is a code search and navigation tool aimed at making sense of large projects like Firefox. It supports full-text and regex searches as well as structural queries like “Find all the callers of this function.” Behind the scenes, it uses trigram indices, elasticsearch, and static analysis data collected by instrumented compilers to make searches faster and more accurate than is possible with simple tools like grep. DXR also exposes a plugin API through which understanding of more languages can be added. Here’s an example of DXR running against the Firefox codebase. It looks like this: Contents 1 DXR Documentation, Release 2.0 2 Contents CHAPTER 1 Contents 1.1 Welcome To The DXR Community Though DXR got its start at Mozilla, it’s seen contributions from a variety of companies and individuals around the world. We welcome contributions of code, bug reports, and helpful feedback. 1.1.1 Bug Reports Did something explode? Not act as you expected? Let us know. 1.1.2 Submitting Patches To contribute code, just file a pull request. Include tests to double your chances of getting it merged and qualify for a free Bundt cake.