A Humanized Yeast Phenomic Model of Deoxycytidine Kinase to Predict Genetic Buffering of Nucleoside Analog Cytotoxicity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Screening and Identification of Key Biomarkers in Clear Cell Renal Cell Carcinoma Based on Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Screening and identification of key biomarkers in clear cell renal cell carcinoma based on bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Clear cell renal cell carcinoma (ccRCC) is one of the most common types of malignancy of the urinary system. The pathogenesis and effective diagnosis of ccRCC have become popular topics for research in the previous decade. In the current study, an integrated bioinformatics analysis was performed to identify core genes associated in ccRCC. An expression dataset (GSE105261) was downloaded from the Gene Expression Omnibus database, and included 26 ccRCC and 9 normal kideny samples. Assessment of the microarray dataset led to the recognition of differentially expressed genes (DEGs), which was subsequently used for pathway and gene ontology (GO) enrichment analysis. -

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

Mapping Influenza-Induced Posttranslational Modifications On
viruses Article Mapping Influenza-Induced Posttranslational Modifications on Histones from CD8+ T Cells Svetlana Rezinciuc 1, Zhixin Tian 2, Si Wu 2, Shawna Hengel 2, Ljiljana Pasa-Tolic 2 and Heather S. Smallwood 1,3,* 1 Department of Pediatrics, University of Tennessee Health Science Center, Memphis, TN 38163, USA; [email protected] 2 Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, WA 99354, USA; [email protected] (Z.T.); [email protected] (S.W.); [email protected] (S.H.); [email protected] (L.P.-T.) 3 Children’s Foundation Research Institute, Memphis, TN 38105, USA * Correspondence: [email protected]; Tel.: +1-(901)-448–3068 Academic Editor: Italo Tempera Received: 10 October 2020; Accepted: 2 December 2020; Published: 8 December 2020 Abstract: T cell function is determined by transcriptional networks that are regulated by epigenetic programming via posttranslational modifications (PTMs) to histone proteins and DNA. Bottom-up mass spectrometry (MS) can identify histone PTMs, whereas intact protein analysis by MS can detect species missed by bottom-up approaches. We used a novel approach of online two-dimensional liquid chromatography-tandem MS with high-resolution reversed-phase liquid chromatography (RPLC), alternating electron transfer dissociation (ETD) and collision-induced dissociation (CID) on precursor ions to maximize fragmentation of uniquely modified species. The first online RPLC separation sorted histone families, then RPLC or weak cation exchange hydrophilic interaction liquid chromatography (WCX-HILIC) separated species heavily clad in PTMs. Tentative identifications were assigned by matching proteoform masses to predicted theoretical masses that were verified with tandem MS. We used this innovative approach for histone-intact protein PTM mapping (HiPTMap) to identify and quantify proteoforms purified from CD8 T cells after in vivo influenza infection. -
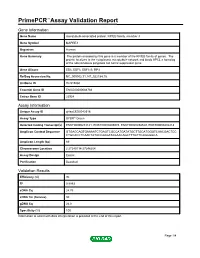
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name microtubule-associated protein, RP/EB family, member 3 Gene Symbol MAPRE3 Organism Human Gene Summary The protein encoded by this gene is a member of the RP/EB family of genes. The protein localizes to the cytoplasmic microtubule network and binds APCL a homolog of the adenomatous polyposis coli tumor suppressor gene. Gene Aliases EB3, EBF3, EBF3-S, RP3 RefSeq Accession No. NC_000002.11, NT_022184.15 UniGene ID Hs.515860 Ensembl Gene ID ENSG00000084764 Entrez Gene ID 22924 Assay Information Unique Assay ID qHsaCED0042518 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000233121, ENST00000405074, ENST00000458529, ENST00000402218 Amplicon Context Sequence GTGACCAGTGAAAATCTGAGTCGCCATGATATGCTTGCATGGGTCAACGACTCC CTGCACCTCAACTATACCAAGATAGAACAGCTTTGTTCAGGGGCA Amplicon Length (bp) 69 Chromosome Location 2:27245114-27246204 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 90 R2 0.9993 cDNA Cq 24.76 cDNA Tm (Celsius) 80 gDNA Cq 26.8 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report MAPRE3, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Human Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -

Molecular Clock Is Involved in Predictive Circadian Adjustment of Renal Function
Molecular clock is involved in predictive circadian adjustment of renal function Annie Mercier Zubera,1,2, Gabriel Centenoa,2, Sylvain Pradervandb, Svetlana Nikolaevaa,c, Lionel Maquelina, Le´ onard Cardinauxa, Olivier Bonnya,d, and Dmitri Firsova,3 aDepartment of Pharmacology and Toxicology, University of Lausanne, 1005 Lausanne, Switzerland; bDNA Array Facility, University of Lausanne, 1015 Lausanne, Switzerland; cInstitute of Evolutionary Physiology and Biochemistry, 194223 St-Petersburg, Russia; and dService of Nephrology, Lausanne University Hospital, 1005 Lausanne, Switzerland Edited by Maurice B. Burg, National Heart, Lung, and Blood Institute, Bethesda, MD, and approved August 6, 2009 (received for review May 4, 2009) Renal excretion of water and major electrolytes exhibits a significant anticipate upcoming circadian environmental challenges (activity, circadian rhythm. This functional periodicity is believed to result, at feeding, etc). least in part, from circadian changes in secretion/reabsorption capac- The most obvious manifestation of circadian rhythmicity of renal ities of the distal nephron and collecting ducts. Here, we studied the function is a well-marked difference in the volume of urine forma- molecular mechanisms underlying circadian rhythms in the distal tion/excretion between the day and the night. The urinary excretion nephron segments, i.e., distal convoluted tubule (DCT) and connect- ϩ ϩ Ϫ Ϫ 2ϩ 2ϩ of all major solutes (Na ,K ,Cl , urea, PO4 ,Ca ,Mg )also ing tubule (CNT) and the cortical collecting duct (CCD). Temporal follows a circadian oscillating pattern. Although, renal excretion expression analysis performed on microdissected mouse DCT/CNT or rhythms are apparently synchronized with circadian rhythms of CCD revealed a marked circadian rhythmicity in the expression of a activity/feeding, they have been shown to persist over long periods large number of genes crucially involved in various homeostatic of time under experimental conditions in which external factors functions of the kidney. -

In Silico Prediction of High-Resolution Hi-C Interaction Matrices
ARTICLE https://doi.org/10.1038/s41467-019-13423-8 OPEN In silico prediction of high-resolution Hi-C interaction matrices Shilu Zhang1, Deborah Chasman 1, Sara Knaack1 & Sushmita Roy1,2* The three-dimensional (3D) organization of the genome plays an important role in gene regulation bringing distal sequence elements in 3D proximity to genes hundreds of kilobases away. Hi-C is a powerful genome-wide technique to study 3D genome organization. Owing to 1234567890():,; experimental costs, high resolution Hi-C datasets are limited to a few cell lines. Computa- tional prediction of Hi-C counts can offer a scalable and inexpensive approach to examine 3D genome organization across multiple cellular contexts. Here we present HiC-Reg, an approach to predict contact counts from one-dimensional regulatory signals. HiC-Reg pre- dictions identify topologically associating domains and significant interactions that are enri- ched for CCCTC-binding factor (CTCF) bidirectional motifs and interactions identified from complementary sources. CTCF and chromatin marks, especially repressive and elongation marks, are most important for HiC-Reg’s predictive performance. Taken together, HiC-Reg provides a powerful framework to generate high-resolution profiles of contact counts that can be used to study individual locus level interactions and higher-order organizational units of the genome. 1 Wisconsin Institute for Discovery, 330 North Orchard Street, Madison, WI 53715, USA. 2 Department of Biostatistics and Medical Informatics, University of Wisconsin-Madison, Madison, WI 53715, USA. *email: [email protected] NATURE COMMUNICATIONS | (2019) 10:5449 | https://doi.org/10.1038/s41467-019-13423-8 | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-019-13423-8 he three-dimensional (3D) organization of the genome has Results Temerged as an important component of the gene regulation HiC-Reg for predicting contact count using Random Forests. -

The Microtubule-Associated Protein MAPRE2 Is Involved in Perineural Invasion of Pancreatic Cancer Cells
1111-1116.qxd 8/9/2009 08:18 Ì ™ÂÏ›‰·1111 INTERNATIONAL JOURNAL OF ONCOLOGY 35: 1111-1116, 2009 The microtubule-associated protein MAPRE2 is involved in perineural invasion of pancreatic cancer cells IVANE ABIATARI1*, SONJA GILLEN1*, TIAGO DeOLIVEIRA1, THERESA KLOSE1, KONG BO1, NATHALIA A. GIESE2, HELMUT FRIESS1 and JÖRG KLEEFF1,3 1Department of General Surgery, Technische Universität München, Munich; 2Department of General Surgery, University of Heidelberg, Heidelberg, Germany; 3Center of Cancer Systems Biology, Department of Medicine, Caritas St. Elizabeth's Medical Center, Tufts University School of Medicine, Boston, MA 02135-2997, USA Received May 4, 2009; Accepted July 10, 2009 DOI: 10.3892/ijo_00000426 Abstract. Perineural invasion of tumor cells is a characteristic Defining molecular mechanisms that allow pancreatic cancer feature of human pancreatic cancer. Unrevealing the molecular cells to grow along and into nerves is therefore important for mechanisms that enable cancer cells to invade and grow along the development of novel therapeutic strategies in pancreatic nerves is important for the development of novel therapeutic cancer, a disease whose incidence virtually mirrors its mortality strategies in this disease. We have previously identified tran- rate (5). We have previously identified and described in detail scriptional changes in highly nerve invasive pancreatic cancer the transcriptome signature of perineural invasion in pancreatic cells. Here we further analyzed one of the identified de- cancer by generating highly nerve invasive pancreatic cancer regulated genes, MAPRE2, a microtubule-associated protein. cells (6). One of the genes that displayed a significant up- MAPRE2 expression was significantly increased in high versus regulation in pancreatic cancer cells with a high potential for less nerve invasive pancreatic cancer cells, and changes of nerve invasion was MAPRE2. -

The Roles of Histone Deacetylase 5 and the Histone Methyltransferase Adaptor WDR5 in Myc Oncogenesis
The Roles of Histone Deacetylase 5 and the Histone Methyltransferase Adaptor WDR5 in Myc oncogenesis By Yuting Sun This thesis is submitted in fulfilment of the requirements for the degree of Doctor of Philosophy at the University of New South Wales Children’s Cancer Institute Australia for Medical Research School of Women’s and Children’s Health, Faculty of Medicine University of New South Wales Australia August 2014 PLEASE TYPE THE UNIVERSITY OF NEW SOUTH WALES Thesis/Dissertation Sheet Surname or Family name: Sun First name: Yuting Other name/s: Abbreviation for degree as given in the University calendar: PhD School : School of·Women's and Children's Health Faculty: Faculty of Medicine Title: The Roles of Histone Deacetylase 5 and the Histone Methyltransferase Adaptor WDR5 in Myc oncogenesis. Abstract 350 words maximum: (PLEASE TYPE) N-Myc Induces neuroblastoma by regulating the expression of target genes and proteins, and N-Myc protein is degraded by Fbxw7 and NEDD4 and stabilized by Aurora A. The class lla histone deacetylase HDAC5 suppresses gene transcription, and blocks myoblast and leukaemia cell differentiation. While histone H3 lysine 4 (H3K4) trimethylation at target gene promoters is a pre-requisite for Myc· induced transcriptional activation, WDRS, as a histone H3K4 methyltransferase presenter, is required for H3K4 methylation and transcriptional activation mediated by a histone H3K4 methyltransferase complex. Here, I investigated the roles of HDAC5 and WDR5 in N-Myc overexpressing neuroblastoma. I have found that N-Myc upregulates HDAC5 protein expression, and that HDAC5 represses NEDD4 gene expression, increases Aurora A gene expression and consequently upregulates N-Myc protein expression in neuroblastoma cells. -

Determining HDAC8 Substrate Specificity by Noah Ariel Wolfson A
Determining HDAC8 substrate specificity by Noah Ariel Wolfson A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Biological Chemistry) in the University of Michigan 2014 Doctoral Committee: Professor Carol A. Fierke, Chair Professor Robert S. Fuller Professor Anna K. Mapp Associate Professor Patrick J. O’Brien Associate Professor Raymond C. Trievel Dedication My thesis is dedicated to all my family, mentors, and friends who made getting to this point possible. ii Table of Contents Dedication ....................................................................................................................................... ii List of Figures .............................................................................................................................. viii List of Tables .................................................................................................................................. x List of Appendices ......................................................................................................................... xi Abstract ......................................................................................................................................... xii Chapter 1 HDAC8 substrates: Histones and beyond ...................................................................... 1 Overview ..................................................................................................................................... 1 HDAC introduction -

Identification of Conserved Genes Triggering Puberty in European Sea
Blázquez et al. BMC Genomics (2017) 18:441 DOI 10.1186/s12864-017-3823-2 RESEARCHARTICLE Open Access Identification of conserved genes triggering puberty in European sea bass males (Dicentrarchus labrax) by microarray expression profiling Mercedes Blázquez1,2* , Paula Medina1,2,3, Berta Crespo1,4, Ana Gómez1 and Silvia Zanuy1* Abstract Background: Spermatogenesisisacomplexprocesscharacterized by the activation and/or repression of a number of genes in a spatio-temporal manner. Pubertal development in males starts with the onset of the first spermatogenesis and implies the division of primary spermatogonia and their subsequent entry into meiosis. This study is aimed at the characterization of genes involved in the onset of puberty in European sea bass, and constitutes the first transcriptomic approach focused on meiosis in this species. Results: European sea bass testes collected at the onset of puberty (first successful reproduction) were grouped in stage I (resting stage), and stage II (proliferative stage). Transition from stage I to stage II was marked by an increase of 11ketotestosterone (11KT), the main fish androgen, whereas the transcriptomic study resulted in 315 genes differentially expressed between the two stages. The onset of puberty induced 1) an up-regulation of genes involved in cell proliferation, cell cycle and meiosis progression, 2) changes in genes related with reproduction and growth, and 3) a down-regulation of genes included in the retinoic acid (RA) signalling pathway. The analysis of GO-terms and biological pathways showed that cell cycle, cell division, cellular metabolic processes, and reproduction were affected, consistent with the early events that occur during the onset of puberty. -

Screening and Identification of Hub Genes in Bladder Cancer by Bioinformatics Analysis and KIF11 Is a Potential Prognostic Biomarker
ONCOLOGY LETTERS 21: 205, 2021 Screening and identification of hub genes in bladder cancer by bioinformatics analysis and KIF11 is a potential prognostic biomarker XIAO‑CONG MO1,2*, ZI‑TONG ZHANG1,3*, MENG‑JIA SONG1,2, ZI‑QI ZHOU1,2, JIAN‑XIONG ZENG1,2, YU‑FEI DU1,2, FENG‑ZE SUN1,2, JIE‑YING YANG1,2, JUN‑YI HE1,2, YUE HUANG1,2, JIAN‑CHUAN XIA1,2 and DE‑SHENG WENG1,2 1State Key Laboratory of Oncology in South China, Collaborative Innovation Centre for Cancer Medicine; 2Department of Biotherapy, Sun Yat‑Sen University Cancer Center; 3Department of Radiation Oncology, Sun Yat‑Sen University Cancer Center, Guangzhou, Guangdong 510060, P.R. China Received July 31, 2020; Accepted December 18, 2020 DOI: 10.3892/ol.2021.12466 Abstract. Bladder cancer (BC) is the ninth most common immunohistochemistry and western blotting. In summary, lethal malignancy worldwide. Great efforts have been devoted KIF11 was significantly upregulated in BC and might act as to clarify the pathogenesis of BC, but the underlying molecular a potential prognostic biomarker. The present identification mechanisms remain unclear. To screen for the genes associated of DEGs and hub genes in BC may provide novel insight for with the progression and carcinogenesis of BC, three datasets investigating the molecular mechanisms of BC. were obtained from the Gene Expression Omnibus. A total of 37 tumor and 16 non‑cancerous samples were analyzed to Introduction identify differentially expressed genes (DEGs). Subsequently, 141 genes were identified, including 55 upregulated and Bladder cancer (BC) is the ninth most common malignancy 86 downregulated genes. The protein‑protein interaction worldwide with substantial morbidity and mortality. -

Genetic Drivers of Pancreatic Islet Function
| INVESTIGATION Genetic Drivers of Pancreatic Islet Function Mark P. Keller,*,1 Daniel M. Gatti,†,1 Kathryn L. Schueler,* Mary E. Rabaglia,* Donnie S. Stapleton,* Petr Simecek,† Matthew Vincent,† Sadie Allen,‡ Aimee Teo Broman,§ Rhonda Bacher,§ Christina Kendziorski,§ Karl W. Broman,§ Brian S. Yandell,** Gary A. Churchill,†,2 and Alan D. Attie*,2 *Department of Biochemistry, §Department of Biostatistics and Medical Informatics, and **Department of Horticulture, University of Wisconsin–Madison, Wisconsin 53706-1544, †The Jackson Laboratory, Bar Harbor, Maine 06409, and ‡Maine School of Science and Mathematics, Limestone, Maine 06409, ORCID IDs: 0000-0002-7405-5552 (M.P.K.); 0000-0002-4914-6671 (K.W.B.); 0000-0001-9190-9284 (G.A.C.); 0000-0002-0568-2261 (A.D.A.) ABSTRACT The majority of gene loci that have been associated with type 2 diabetes play a role in pancreatic islet function. To evaluate the role of islet gene expression in the etiology of diabetes, we sensitized a genetically diverse mouse population with a Western diet high in fat (45% kcal) and sucrose (34%) and carried out genome-wide association mapping of diabetes-related phenotypes. We quantified mRNA abundance in the islets and identified 18,820 expression QTL. We applied mediation analysis to identify candidate causal driver genes at loci that affect the abundance of numerous transcripts. These include two genes previously associated with monogenic diabetes (PDX1 and HNF4A), as well as three genes with nominal association with diabetes-related traits in humans (FAM83E, IL6ST, and SAT2). We grouped transcripts into gene modules and mapped regulatory loci for modules enriched with transcripts specific for a-cells, and another specific for d-cells.