Copyright © 2019 by Yue Wu
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 2 2. Q Labelling in Hyoscyamine
Durham E-Theses The biosynthesis of the tropane alkaloid hyoscyamine in datura stramonium Wong, Chi W. How to cite: Wong, Chi W. (1999) The biosynthesis of the tropane alkaloid hyoscyamine in datura stramonium, Durham theses, Durham University. Available at Durham E-Theses Online: http://etheses.dur.ac.uk/4310/ Use policy The full-text may be used and/or reproduced, and given to third parties in any format or medium, without prior permission or charge, for personal research or study, educational, or not-for-prot purposes provided that: • a full bibliographic reference is made to the original source • a link is made to the metadata record in Durham E-Theses • the full-text is not changed in any way The full-text must not be sold in any format or medium without the formal permission of the copyright holders. Please consult the full Durham E-Theses policy for further details. Academic Support Oce, Durham University, University Oce, Old Elvet, Durham DH1 3HP e-mail: [email protected] Tel: +44 0191 334 6107 http://etheses.dur.ac.uk COPYRIGHT The copyright of this thesis rests with the author. No quotation form it should be published without prior consent, and any information derived from this thesis should be acknowledged. DECLARATION The work contained in this thesis was carried out in the Department of Chemistry at the University of Durham between October 1995 and September 1998. All the work was carried out by the author, unless otherwise indicated. It has not been previously submitted for a degree at this or any other university. -

The Effect of Overfeeding and Obesity on Canine Adipose Tissue and Skeletal Muscle Transcriptomes by Ryan Walter Grant Disserta
THE EFFECT OF OVERFEEDING AND OBESITY ON CANINE ADIPOSE TISSUE AND SKELETAL MUSCLE TRANSCRIPTOMES BY RYAN WALTER GRANT DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Nutritional Sciences in the Graduate College of the University of Illinois at Urbana-Champaign, 2011 Urbana, Illinois Doctoral Committee: Professor Sharon M. Donovan, Chair Associate Professor Kelly S. Swanson, Director of Research Associate Professor Thomas K. Graves Associate Professor Manabu T. Nakamura Abstract Overweight dogs have a reduced life expectancy and increased risk of chronic disease. During obesity development, adipose tissue undergoes major expansion and remodeling, but the biological processes involved are not well understood. The objective of study 1 was to analyze global gene expression profiles of adipose tissue in dogs, fed a high-fat (47% kcal/g) diet, during the transition from a lean to obese phenotype. Nine female beagles were randomized to ad libitum (n=5) feeding or body weight maintenance (n=4). Subcutaneous adipose tissue biopsy, skeletal muscle biopsy and blood samples were collected, and dual x-ray absorptiometry measurements were taken at 0, 4, 8, 12, and 24 wk of feeding. Ad libitum feeding increased (P<0.05) body weight, body fat mass, adipocyte size and serum leptin concentrations. Microarrays displayed 1,665 differentially expressed genes in adipose tissue over time in the ad libitum fed dogs. Alterations were observed in many homeostatic processes including metabolism, oxidative stress, mitochondrial homeostasis, and extracellular matrix. Our data implies that during obesity development subcutaneous adipose tissue has a large capacity for expansion, which is accompanied by tissue remodeling and short-term adaptations to the metabolic stresses associated with ad libitum feeding. -

(12) Patent Application Publication (10) Pub. No.: US 2009/0269772 A1 Califano Et Al
US 20090269772A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2009/0269772 A1 Califano et al. (43) Pub. Date: Oct. 29, 2009 (54) SYSTEMS AND METHODS FOR Publication Classification IDENTIFYING COMBINATIONS OF (51) Int. Cl. COMPOUNDS OF THERAPEUTIC INTEREST CI2O I/68 (2006.01) CI2O 1/02 (2006.01) (76) Inventors: Andrea Califano, New York, NY G06N 5/02 (2006.01) (US); Riccardo Dalla-Favera, New (52) U.S. Cl. ........... 435/6: 435/29: 706/54; 707/E17.014 York, NY (US); Owen A. (57) ABSTRACT O'Connor, New York, NY (US) Systems, methods, and apparatus for searching for a combi nation of compounds of therapeutic interest are provided. Correspondence Address: Cell-based assays are performed, each cell-based assay JONES DAY exposing a different sample of cells to a different compound 222 EAST 41ST ST in a plurality of compounds. From the cell-based assays, a NEW YORK, NY 10017 (US) Subset of the tested compounds is selected. For each respec tive compound in the Subset, a molecular abundance profile from cells exposed to the respective compound is measured. (21) Appl. No.: 12/432,579 Targets of transcription factors and post-translational modu lators of transcription factor activity are inferred from the (22) Filed: Apr. 29, 2009 molecular abundance profile data using information theoretic measures. This data is used to construct an interaction net Related U.S. Application Data work. Variances in edges in the interaction network are used to determine the drug activity profile of compounds in the (60) Provisional application No. 61/048.875, filed on Apr. -

CREB-Mediated Enhancement of Hippocampus-Dependent Memory Consolidation and Reconsolidation
CREB-mediated enhancement of hippocampus-dependent memory consolidation and reconsolidation by Melanie Jay Sekeres A thesis submitted in conformity with the requirements for the degree of Doctorate of Philosophy (PhD) Department of Physiology University of Toronto © Copyright by Melanie Jay Sekeres, 2012 CREB-mediated enhancement of hippocampus-dependent memory consolidation and reconsolidation Melanie Jay Sekeres PhD Department of Physiology University of Toronto 2012 Abstract Memory stabilization following encoding (synaptic consolidation) or memory reactivation (reconsolidation) requires gene expression and protein synthesis. Although consolidation and reconsolidation may be mediated by distinct molecular mechanisms, disrupting the function of the transcription factor CREB (cAMP responsive element binding protein) impairs both processes. We use a gain-of-function approach to show that CREB (and CREB-coactivator CRTC1) can facilitate both synaptic and systems consolidation and reconsolidation. We first examine whether acutely increasing CREB levels in the dorsal hippocampus is sufficient to enhance spatial memory formation in the watermaze. Locally and acutely increasing CREB in the dorsal hippocampus using viral vectors is sufficient to induce robust spatial memory in two conditions which do not normally support consolidation, weakly-trained wild-type (WT) mice and strongly-trained mutant mice with brain-wide disrupted CREB function. ii CRTCs (CREB regulated transcription co-activators) are a powerful co-activator of CREB, but their role in memory is virtually unexplored. We show, for the first time, that the novel CREB co-activator CRTC1 enhances memory consolidation. Locally increasing CRTC1 (or CREB) in the dorsal hippocampus of WT mice prior to weak context fear conditioning facilitates consolidation of precise context memory. -

All Enzymes in BRENDA™ the Comprehensive Enzyme Information System
All enzymes in BRENDA™ The Comprehensive Enzyme Information System http://www.brenda-enzymes.org/index.php4?page=information/all_enzymes.php4 1.1.1.1 alcohol dehydrogenase 1.1.1.B1 D-arabitol-phosphate dehydrogenase 1.1.1.2 alcohol dehydrogenase (NADP+) 1.1.1.B3 (S)-specific secondary alcohol dehydrogenase 1.1.1.3 homoserine dehydrogenase 1.1.1.B4 (R)-specific secondary alcohol dehydrogenase 1.1.1.4 (R,R)-butanediol dehydrogenase 1.1.1.5 acetoin dehydrogenase 1.1.1.B5 NADP-retinol dehydrogenase 1.1.1.6 glycerol dehydrogenase 1.1.1.7 propanediol-phosphate dehydrogenase 1.1.1.8 glycerol-3-phosphate dehydrogenase (NAD+) 1.1.1.9 D-xylulose reductase 1.1.1.10 L-xylulose reductase 1.1.1.11 D-arabinitol 4-dehydrogenase 1.1.1.12 L-arabinitol 4-dehydrogenase 1.1.1.13 L-arabinitol 2-dehydrogenase 1.1.1.14 L-iditol 2-dehydrogenase 1.1.1.15 D-iditol 2-dehydrogenase 1.1.1.16 galactitol 2-dehydrogenase 1.1.1.17 mannitol-1-phosphate 5-dehydrogenase 1.1.1.18 inositol 2-dehydrogenase 1.1.1.19 glucuronate reductase 1.1.1.20 glucuronolactone reductase 1.1.1.21 aldehyde reductase 1.1.1.22 UDP-glucose 6-dehydrogenase 1.1.1.23 histidinol dehydrogenase 1.1.1.24 quinate dehydrogenase 1.1.1.25 shikimate dehydrogenase 1.1.1.26 glyoxylate reductase 1.1.1.27 L-lactate dehydrogenase 1.1.1.28 D-lactate dehydrogenase 1.1.1.29 glycerate dehydrogenase 1.1.1.30 3-hydroxybutyrate dehydrogenase 1.1.1.31 3-hydroxyisobutyrate dehydrogenase 1.1.1.32 mevaldate reductase 1.1.1.33 mevaldate reductase (NADPH) 1.1.1.34 hydroxymethylglutaryl-CoA reductase (NADPH) 1.1.1.35 3-hydroxyacyl-CoA -

Durham E-Theses
Durham E-Theses The biosynthesis of the tropane alkaloid hyoscyamine in datura stramonium Wong, Chi W. How to cite: Wong, Chi W. (1999) The biosynthesis of the tropane alkaloid hyoscyamine in datura stramonium, Durham theses, Durham University. Available at Durham E-Theses Online: http://etheses.dur.ac.uk/4310/ Use policy The full-text may be used and/or reproduced, and given to third parties in any format or medium, without prior permission or charge, for personal research or study, educational, or not-for-prot purposes provided that: • a full bibliographic reference is made to the original source • a link is made to the metadata record in Durham E-Theses • the full-text is not changed in any way The full-text must not be sold in any format or medium without the formal permission of the copyright holders. Please consult the full Durham E-Theses policy for further details. Academic Support Oce, Durham University, University Oce, Old Elvet, Durham DH1 3HP e-mail: [email protected] Tel: +44 0191 334 6107 http://etheses.dur.ac.uk COPYRIGHT The copyright of this thesis rests with the author. No quotation form it should be published without prior consent, and any information derived from this thesis should be acknowledged. DECLARATION The work contained in this thesis was carried out in the Department of Chemistry at the University of Durham between October 1995 and September 1998. All the work was carried out by the author, unless otherwise indicated. It has not been previously submitted for a degree at this or any other university. -
Co-Activator Candidate Interactions for Orphan Nuclear Receptor NR2E1 Ximena Corso-Díaz1,2, Charles N
Corso-Díaz et al. BMC Genomics (2016) 17:832 DOI 10.1186/s12864-016-3173-5 RESEARCH ARTICLE Open Access Co-activator candidate interactions for orphan nuclear receptor NR2E1 Ximena Corso-Díaz1,2, Charles N. de Leeuw1,3, Vivian Alonso1, Diana Melchers4, Bibiana K. Y. Wong1,3, René Houtman4 and Elizabeth M. Simpson1,2,3,5,6* Abstract Background: NR2E1 (Tlx) is an orphan nuclear receptor that regulates the maintenance and self-renewal of neural stem cells, and promotes tumourigenesis. Nr2e1-null mice exhibit reduced cortical and limbic structures and pronounced retinal dystrophy. NR2E1 functions mainly as a repressor of gene transcription in association with the co-repressors atrophin-1, LSD1, HDAC and BCL11A. Recent evidence suggests that NR2E1 also acts as an activator of gene transcription. However, co-activator complexes that interact with NR2E1 have not yet been identified. In order to find potential novel co-regulators for NR2E1, we used a microarray assay for real-time analysis of co-regulator– nuclear receptor interaction (MARCoNI) that contains peptides representing interaction motifs from potential co- regulatory proteins, including known co-activator nuclear receptor box sequences (LxxLL motif). Results: We found that NR2E1 binds strongly to an atrophin-1 peptide (Atro box) used as positive control and to 19 other peptides that constitute candidate NR2E1 partners. Two of these proteins, p300 and androgen receptor (AR), were further validated by reciprocal pull-down assays. The specificity of NR2E1 binding to peptides in the array was evaluated using two single amino acid variants, R274G and R276Q, which disrupted the majority of the binding interactions observed with wild-type NR2E1. -

Integrated Physiology—Insulin Secretion in Vivo
INTEGRATED PHYSIOLOGY—INSULINCATEGORY SECRETION IN VIVO diet rich in carbohydrate (CHO), b) diet rich in monounsaturated fat (MUFA) INTEGRATED PHYSIOLOGY—INSULIN SECRETION and c) diet high in saturated fat (SAT). Total cholesterol, LDL-C and ApoB IN VIVO levels were increase after SAT diets as compared with MUFA and CHO diets. PAT from 9 subjects was collected and Microarrays RT–qPCR was used for transcriptomic analysis of 86 GEN-FAT. The table shows relationship 2585-PO between diet induced changes in GEN-FAT expression and lipid profi le. In Age as a Modifi er of β-Cell Response to Insulin Resistance among summary, specifi c gene-expression on GEN-FAT in peripheral adipose tissue Normo glycemic African American and Caucasian Offspring of Parents are related with the specifi c plasma lipid profi le diet-induced by different with or without Diabetes macronutrient composition. EBENEZER NYENWE, CHIMAROKE EDEOGA, EMMANUEL CHAPP-JUMBO, JIM WAN, SAMUEL DAGOGO-JACK, Memphis, TN Table 1. Correlationship between changes on GEN of Fatty Acid Metabolism We studied the interaction of ethnicity, age, insulin action and secretion and Lipid profi le after the three dietary. among normoglycemic African-Americans (AA) and Caucasians who were Correlationship, ( n=9) GEN-FAT MUFA SAT CHO offspring of parents with or without type 2 diabetes (T2D). Given the genetic basis of T2D, we postulated that normoglycemic offspring of T2D Free Fatty acids FADS1 -0.43; p=0.24 -0.25; p=0.52 -0.88; p=0.002 ACSL6 0.85; p=0.001 0.85; p=0.004 -0.23; p=0.54 parents would exhibit discernable glucoregulatory defects. -

FARE2019 WINNERS Sorted by Institute
FARE2019 WINNERS Sorted By Institute Ling Zhang Postdoctoral Fellow CC Radiology/Imaging/PET and Neuroimaging Convolutional Invasion and Expansion Networks for Tumor Growth Prediction This study investigates the possibility of using deep learning to model and predict tumor growth, which has long been formulated by mathematical modeling. Tumor growth is associated with two fundamental processes: cell invasion and mass-effect. Medical imaging data provides non-invasive and in vivo- measurements of these underlying tumor physiological parameters over time. We propose a two- stream end-to-end convolutional neural network (ConvNet) to directly represent and learn the cell invasion and mass-effect from longitudinal medical images, and to predict the subsequent involvement regions of a tumor. The network architecture can easily be trained on population data and personalized to a target patient, unlike most previous mathematical modeling methods that fail to incorporate population data. Experiments are taken on a longitudinal pancreatic tumor data set with comparison with a state-of-the-art mathematical model-based approach. To the best of our knowledge, this is the first time to use learnable ConvNet models for explicitly capturing the two fundamental processes of tumor growth. The invasion network can make its prediction based on the metabolic rate, cell density and tumor boundary, all derived from the multi-model imaging data. Mass-effect â the mechanical force exerted by the growing tumor â can be approximated by the expansion/shrink motion (magnitude and orientation) of the tumor mass. This expansion/shrink cue is captured by optical flow computing, based on which the expansion network is trained to infer tumor growth. -
Zhongqi Chen
PHYSIOLOGICAL, TRANSCRIPTOMIC AND GENOMIC MECHANISMS OF THERMAL ADAPTATION IN ONCORHYNCHUS MYKISS by Zhongqi Chen B.Sc., Xuzhou Normal University, 2007 M.Sc., Northwest A&F University, 2010 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in THE FACULTY OF GRADUATE AND POSTDOCTORAL STUDIES (Zoology) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) January 2017 © Zhongqi Chen, 2017 Abstract Given the rate and magnitude of the ongoing global warming, there is some urgency to understand the underlying mechanism of thermal adaptation to evaluate and predict the ecological consequences. This thesis used Oncorhynchus mykiss from different thermal regimes to examine thermal adaptation at physiological, transcriptomic and genomic levels. Thermal tolerance was examined in three redband trout (O. mykiss gairdneri) populations from warm desert and cool montane climates (Idaho, USA), as well as in a domesticated rainbow trout (O. mykiss) strain raised in a thermally challenging environment for over 19 generations (Western Australia, Australia). Acclimated to 15°C, the desert redband trout had the highest critical thermal maximum (CTMAX; 29.7°C) and maintained an almost constant absolute aerobic scope (AAS) across a broader range of test temperatures (12-24°C) than seen in other strains, but had the lowest peak AAS, suggesting a tradeoff between thermal performance and tolerance. Western Australian rainbow trout had the highest AAS, even when tested at 21°C, which may be a result of hatchery selection for thermal performance. Although the rate transition temperatures for maximum heart rate (Arrhenius breakpoint and arrhythmia temperature for fH,max) were similar among all populations, fH,max was the highest in the desert redband trout population at all temperatures. -
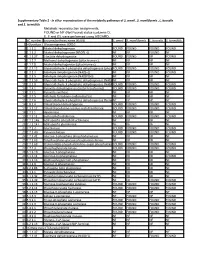
Supplementary Table 2 - in Silico Reconstruction of the Metabolic Pathways of S
Supplementary Table 2 - In silico reconstruction of the metabolic pathways of S. amnii , S. moniliformis , L. buccalis and S. termiditis Metabolic reconstruction assignments, FOUND or NF (Not Found) status (columns D, E, F and G), were performed using ASGARD, EC number Enzyme/pathway name (KEGG) S. amnii S. moniliformis L. buccalis S. termiditis 1 >Glycolysis / Gluconeogenesis 00010 2 1.1.1.1 Alcohol dehydrogenase. FOUND FOUND FOUND FOUND 3 1.1.1.2 Alcohol dehydrogenase (NADP(+)). NF NF FOUND NF 4 1.1.1.27 L-lactate dehydrogenase. FOUND FOUND NF FOUND 5 1.1.2.7 Methanol dehydrogenase (cytochrome c). NF NF NF NF 6 1.1.2.8 Alcohol dehydrogenase (cytochrome c). NF NF NF NF 7 1.2.1.12 Glyceraldehyde-3-phosphate dehydrogenase (phosphorylating).FOUND FOUND FOUND FOUND 8 1.2.1.3 Aldehyde dehydrogenase (NAD(+)). NF NF FOUND FOUND 9 1.2.1.5 Aldehyde dehydrogenase (NAD(P)(+)). NF NF NF NF 10 1.2.1.59 Glyceraldehyde-3-phosphate dehydrogenase (NAD(P)(+))NF (phosphorylating).NF NF NF 11 1.2.1.9 Glyceraldehyde-3-phosphate dehydrogenase (NADP(+)).FOUND FOUND FOUND FOUND 12 1.2.4.1 Pyruvate dehydrogenase (acetyl-transferring). FOUND FOUND FOUND FOUND 13 1.2.7.1 Pyruvate synthase. NF NF NF NF 14 1.2.7.5 Aldehyde ferredoxin oxidoreductase. NF NF NF NF 15 1.2.7.6 Glyceraldehyde-3-phosphate dehydrogenase (ferredoxin).NF NF NF NF 16 1.8.1.4 Dihydrolipoyl dehydrogenase. FOUND FOUND FOUND FOUND 17 2.3.1.12 Dihydrolipoyllysine-residue acetyltransferase. FOUND FOUND FOUND FOUND 18 2.7.1.1 Hexokinase. -

2011 ADA Posters 1261-2041.Indd
INTEGRATED PHYSIOLOGY—INSULINCATEGORY SECRETION IN VIVO 1622-P 1624-P Hyperglycemia Mediated Accelerates Aging in Vascular Endothelial Unique Cellular Responses of Adult Blood-Derived Endothelial Pro- Cells genitor Cells and Mature Endothelial Cells to High Glucose ROKHSANA MORTUZA, SHALI CHEN, BIAO FENG, SUBRATA CHAKRABARTI, EMILY KEATS, ZIA A. KHAN, London, ON, Canada London, ON, Canada, Lindon, ON, Canada Diabetes leads to dysfunction of selected organ systems, and vascular Glucose-induced endothelial cell dysfunction is a main factor causing endothelial cell (EC) dysfunction and loss is the key initiating and perpetuating tissue damage in the eye, kidney, heart and nerve in Diabetes. We step in the development of secondary diabetic complications. A number hypothesized that hyperglycemia induced oxidative stress causes an of studies have investigated the effect of diabetes on non-vasculogenic accelerated aging process and that such process is mediated through precursor cells (also known as early endothelial progenitor cells). However, a alterations of silent information regulator proteins or sirtuins(SIRTs) through comparative study investigating the effects of high levels of glucose on the epigenetic mechanisms. proliferative/vasculogenic endothelial progenitor cells (EPCs; late EPCs) and We investigated glucose induced aging and alterations of SIRTs in two mature ECs is lacking. We herein determined the response of adult blood- types of endothelial cells, human umbilical vein endothelial cells(HUVECs) derived EPCs and mature ECs to high levels glucose. and human microvascular endothelial cells(HMECs). The cells were grown Mononuclear cells (MNCs) from normal adult blood were successfully and propagated in high glucose (25 mmol, HG) and low glucose(5 mmol, NG).