Thesis Printing Final
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Tanibirumab (CUI C3490677) Add to Cart
5/17/2018 NCI Metathesaurus Contains Exact Match Begins With Name Code Property Relationship Source ALL Advanced Search NCIm Version: 201706 Version 2.8 (using LexEVS 6.5) Home | NCIt Hierarchy | Sources | Help Suggest changes to this concept Tanibirumab (CUI C3490677) Add to Cart Table of Contents Terms & Properties Synonym Details Relationships By Source Terms & Properties Concept Unique Identifier (CUI): C3490677 NCI Thesaurus Code: C102877 (see NCI Thesaurus info) Semantic Type: Immunologic Factor Semantic Type: Amino Acid, Peptide, or Protein Semantic Type: Pharmacologic Substance NCIt Definition: A fully human monoclonal antibody targeting the vascular endothelial growth factor receptor 2 (VEGFR2), with potential antiangiogenic activity. Upon administration, tanibirumab specifically binds to VEGFR2, thereby preventing the binding of its ligand VEGF. This may result in the inhibition of tumor angiogenesis and a decrease in tumor nutrient supply. VEGFR2 is a pro-angiogenic growth factor receptor tyrosine kinase expressed by endothelial cells, while VEGF is overexpressed in many tumors and is correlated to tumor progression. PDQ Definition: A fully human monoclonal antibody targeting the vascular endothelial growth factor receptor 2 (VEGFR2), with potential antiangiogenic activity. Upon administration, tanibirumab specifically binds to VEGFR2, thereby preventing the binding of its ligand VEGF. This may result in the inhibition of tumor angiogenesis and a decrease in tumor nutrient supply. VEGFR2 is a pro-angiogenic growth factor receptor -

Purging and Haemopoietic Progenitor Cell Selection by CD34 Cell Separation
Bone Marrow Transplantation, (1998) 21, 665–671 1998 Stockton Press All rights reserved 0268–3369/98 $12.00 Purging and haemopoietic progenitor cell selection by CD34؉ cell separation ¨ ¨ W Kruger1, M Gruber1, S Hennings1, N Fehse1, B Fehse1, K Gutensohn2, N Kroger1 and AR Zander1 1Bone Marrow Transplantation Unit, 2Blood Transfusion Service, University Hospital Eppendorf, Hamburg, Germany Summary: High-dose chemotherapy supported by autologous stem cell reinfusion for treatment of metastatic and high-risk female breast cancer is currently under investigation in several Tumour cell contamination of autologous peripheral American and European studies.1,2,3 Tumour cell contami- blood stem cell samples (PBSC) and bone marrow (BM) nation of autologous stem cell products has been described is frequent. Enrichment of CD34+ stem cells is a promis- using immunocytochemistry, reverse transcriptase PCR and ing approach to purging tumour cells from autografts cell culture techniques in approximately one third of har- without damaging progenitor cells. Breast cancer cells vests with a range from 0% to 100%.4–7 The clonogenic were seeded (10؊3؊10؊7) into mononuclear cells from and metastatic potential of tumour cells isolated from blood G-CSF-mobilised PBSC and BM harvests from patients and autografts was demonstrated in cell culture assays and without breast cancer. CD34+ cells were enriched from in nude mice.8–10 The metastatic potential of accidentally mixtures either by immunomagnetic separation (Isolex- retransplanted tumour cells could be investigated by gene 50, and MiniMACS) or by biotin-streptavidin immu- marking of autografts prior to infusion followed by the noaffinity columns (Ceprate-LC). CD34؉ cell fractions detection of marked cells after relapse.11 However, due to were determined by FACS, cancer cells were detected the poor transduction efficacy of epithelial cancer cells by immunocytochemically with an anti-pancytokeratin gene transfer, a negative result would not exclude the possi- antibody. -
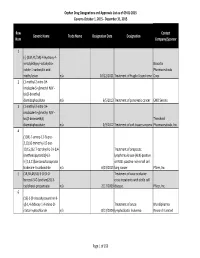
Orphan Drug Designation List
Orphan Drug Designations and Approvals List as of 09‐01‐2015 Governs October 1, 2015 ‐ December 31, 2015 Row Contact Generic Name Trade Name Designation Date Designation Num Company/Sponsor 1 (‐)‐(3aR,4S,7aR)‐4‐Hydroxy‐4‐ m‐tolylethynyl‐octahydro‐ Novartis indole‐1‐carboxylic acid Pharmaceuticals methyl ester n/a 10/12/2011 Treatment of Fragile X syndrome Corp. 2 (1‐methyl‐2‐nitro‐1H‐ imidazole‐5‐yl)methyl N,N'‐ bis(2‐broethyl) diamidophosphate n/a 6/5/2013 Treatment of pancreatic cancer EMD Serono 3 (1‐methyl‐2‐nitro‐1H‐ imidazole‐5‐yl)methyl N,N'‐ bis(2‐bromoethyl) Threshold diamidophosphate n/a 3/9/2012 Treatment of soft tissue sarcoma Pharmaceuticals, Inc. 4 (1OR)‐7‐amino‐12‐fluoro‐ 2,10,16‐trimethyl‐15 oxo‐ 10,15,16,17‐tetrahydro‐2H‐8,4‐ Treatment of anaplastic (metheno)pyrazolo[4,3‐ lymphoma kinase (ALK)‐positive h][2,5,11]benzoxadiazacyclote or ROS1‐positive non‐small cell tradecine‐3‐carbonitrile n/a 6/23/2015 lung cancer Pfizer, Inc. 5 (1R,3R,4R,5S)‐3‐O‐[2‐O‐ Treatment of vaso‐occlusive benzoyl‐3‐O‐(sodium(2S)‐3‐ crisis in patients with sickle cell cyclohexyl‐propanoate‐ n/a 2/17/2009 disease. Pfizer, Inc. 6 (1S)‐1‐(9‐deazahypoxanthin‐9‐ yl)‐1,4‐dideoxy‐1,4‐imino‐D‐ Treatment of acute Mundipharma ribitol‐hydrochloride n/a 8/13/2004 lymphoblastic leukemia Research Limited Page 1 of 359 Orphan Drug Designations and Approvals List as of 09‐01‐2015 Governs October 1, 2015 ‐ December 31, 2015 Row Contact Generic Name Trade Name Designation Date Designation Num Company/Sponsor 7 Treatment of chronic lymphocytic leukemia and related leukemias to include (1S)‐1‐(9‐deazahypoxanthin‐9‐ prolymphocytic leukemia, adult T‐ yl)‐1,4‐dideoxy‐1,4‐imino‐D‐ cell leukemia, and hairy cell Mundipharma ribitol‐hydrochloride n/a 8/10/2004 leukemia Research Ltd. -

(12) Patent Application Publication (10) Pub. No.: US 2009/0226431 A1 Habib (43) Pub
US 20090226431A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2009/0226431 A1 Habib (43) Pub. Date: Sep. 10, 2009 (54) TREATMENT OF CANCER AND OTHER Publication Classification DISEASES (51) Int. Cl. A 6LX 3/575 (2006.01) (76)76) InventorInventor: Nabilabil Habib,Habib. Beirut (LB(LB) C07J 9/00 (2006.01) Correspondence Address: A 6LX 39/395 (2006.01) 101 FEDERAL STREET A6IP 29/00 (2006.01) A6IP35/00 (2006.01) (21) Appl. No.: 12/085,892 A6IP37/00 (2006.01) 1-1. (52) U.S. Cl. ...................... 424/133.1:552/551; 514/182: (22) PCT Filed: Nov.30, 2006 514/171 (86). PCT No.: PCT/US2O06/045665 (57) ABSTRACT .."St. Mar. 6, 2009 The present invention relates to a novel compound (e.g., 24-ethyl-cholestane-3B.5C,6C.-triol), its production, its use, and to methods of treating neoplasms and other tumors as Related U.S. Application Data well as other diseases including hypercholesterolemia, (60) Provisional application No. 60/741,725, filed on Dec. autoimmune diseases, viral diseases (e.g., hepatitis B, hepa 2, 2005. titis C, or HIV), and diabetes. F2: . - 2 . : F2z "..., . Cz: ".. .. 2. , tie - . 2 2. , "Sphagoshgelin , , re Cls Phosphatidiglethanolamine * - 2 .- . t - r y ... CBs .. A . - . Patent Application Publication Sep. 10, 2009 Sheet 1 of 16 US 2009/0226431 A1 E. e'' . Phosphatidylcholine. " . Ez'.. C.2 . Phosphatidylserias. * . - A. z' C. w E. a...2 .". is 2 - - " - B 2. Sphingoshgelin . Cls Phosphatidglethanglamine Figure 1 Patent Application Publication Sep. 10, 2009 Sheet 2 of 16 US 2009/0226431 A1 Chile Phosphater Glycerol Phosphatidylcholine E. -

Lääkealan Turvallisuus- Ja Kehittämiskeskuksen Päätös
Lääkealan turvallisuus- ja kehittämiskeskuksen päätös N:o xxxx lääkeluettelosta Annettu Helsingissä xx päivänä maaliskuuta 2016 ————— Lääkealan turvallisuus- ja kehittämiskeskus on 10 päivänä huhtikuuta 1987 annetun lääke- lain (395/1987) 83 §:n nojalla päättänyt vahvistaa seuraavan lääkeluettelon: 1 § Lääkeaineet ovat valmisteessa suolamuodossa Luettelon tarkoitus teknisen käsiteltävyyden vuoksi. Lääkeaine ja sen suolamuoto ovat biologisesti samanarvoisia. Tämä päätös sisältää luettelon Suomessa lääk- Liitteen 1 A aineet ovat lääkeaineanalogeja ja keellisessä käytössä olevista aineista ja rohdoksis- prohormoneja. Kaikki liitteen 1 A aineet rinnaste- ta. Lääkeluettelo laaditaan ottaen huomioon lää- taan aina vaikutuksen perusteella ainoastaan lää- kelain 3 ja 5 §:n säännökset. kemääräyksellä toimitettaviin lääkkeisiin. Lääkkeellä tarkoitetaan valmistetta tai ainetta, jonka tarkoituksena on sisäisesti tai ulkoisesti 2 § käytettynä parantaa, lievittää tai ehkäistä sairautta Lääkkeitä ovat tai sen oireita ihmisessä tai eläimessä. Lääkkeeksi 1) tämän päätöksen liitteessä 1 luetellut aineet, katsotaan myös sisäisesti tai ulkoisesti käytettävä niiden suolat ja esterit; aine tai aineiden yhdistelmä, jota voidaan käyttää 2) rikoslain 44 luvun 16 §:n 1 momentissa tar- ihmisen tai eläimen elintoimintojen palauttami- koitetuista dopingaineista annetussa valtioneuvos- seksi, korjaamiseksi tai muuttamiseksi farmako- ton asetuksessa kulloinkin luetellut dopingaineet; logisen, immunologisen tai metabolisen vaikutuk- ja sen avulla taikka terveydentilan -

Federal Register / Vol. 60, No. 80 / Wednesday, April 26, 1995 / Notices DIX to the HTSUS—Continued
20558 Federal Register / Vol. 60, No. 80 / Wednesday, April 26, 1995 / Notices DEPARMENT OF THE TREASURY Services, U.S. Customs Service, 1301 TABLE 1.ÐPHARMACEUTICAL APPEN- Constitution Avenue NW, Washington, DIX TO THE HTSUSÐContinued Customs Service D.C. 20229 at (202) 927±1060. CAS No. Pharmaceutical [T.D. 95±33] Dated: April 14, 1995. 52±78±8 ..................... NORETHANDROLONE. A. W. Tennant, 52±86±8 ..................... HALOPERIDOL. Pharmaceutical Tables 1 and 3 of the Director, Office of Laboratories and Scientific 52±88±0 ..................... ATROPINE METHONITRATE. HTSUS 52±90±4 ..................... CYSTEINE. Services. 53±03±2 ..................... PREDNISONE. 53±06±5 ..................... CORTISONE. AGENCY: Customs Service, Department TABLE 1.ÐPHARMACEUTICAL 53±10±1 ..................... HYDROXYDIONE SODIUM SUCCI- of the Treasury. NATE. APPENDIX TO THE HTSUS 53±16±7 ..................... ESTRONE. ACTION: Listing of the products found in 53±18±9 ..................... BIETASERPINE. Table 1 and Table 3 of the CAS No. Pharmaceutical 53±19±0 ..................... MITOTANE. 53±31±6 ..................... MEDIBAZINE. Pharmaceutical Appendix to the N/A ............................. ACTAGARDIN. 53±33±8 ..................... PARAMETHASONE. Harmonized Tariff Schedule of the N/A ............................. ARDACIN. 53±34±9 ..................... FLUPREDNISOLONE. N/A ............................. BICIROMAB. 53±39±4 ..................... OXANDROLONE. United States of America in Chemical N/A ............................. CELUCLORAL. 53±43±0 -

Special Lecture Oxazaphosphorine Cytostatics: Past-Present-Future Seventh Cain Memorial Award Lecture1
[CANCER RESEARCH 49. 1-7. January I, 1989] Special Lecture Oxazaphosphorine Cytostatics: Past-Present-Future Seventh Cain Memorial Award Lecture1 NorbertBrock Department of Cancer Research, ASTA Pharma AG. D-4800 Bielefeld 14. Federal Republic of Germany lin. In the words of Heubner, "I love that word conscientiousness. Abstract It conveys the indispensable association between scientific knowl The development of the oxazaphosphorine cytostatics cyclophos- edge and responsibility—sdentici et conscientia"'. This was one phamide, ifosfamide, and trofosfamide was based on the idea of applying of the essential principles of Wolfgang Heubner at the Pharma the transport form/active form principle to the highly reactive nitrogen cology Institute of the University of Berlin, and I have attempted mustard group. A critical analysis and synopsis of the available results and knowledge will include examination of the extent to which the to base my life and work on this too. Hermann Druckrey was hypotheses on which this concept is based have been confirmed by responsible for my broad introduction to the fundamentals of experimental and clinical findings: pharmacology, and specifically to cancer research. He was one 1. Chemical synthesis succeeded in converting the reactive nitrogen of the founders of the field of oncological pharmacology, the mustard into an inactive transport form (latentiation). scientific basis of which he developed and considerably extended. 2. The requirement that the transport form be enzymatically activated Even in these early years in Berlin we were interested in drugs to to the active form in the target organ (the cancer cell) has been achieved inhibit cell division, and a particularly suitable subject for the by a sequence of metabolic reactions. -

Curriculum Vitae Duan Wang
The Role of Constitutive Androstane Receptor in the Bioactivation of Oxazaphosphorines Item Type dissertation Authors Wang, Duan Publication Date 2013 Abstract Prodrugs are pharmaceutical substances that are administered in an inactive form and are subsequently converted to the active therapeutic moiety through bioactivation. Among them, oxazaphosphorines represent a major class of anti-cancer prodrugs, wit... Keywords constitutive androstane receptor; drug metabolism; oxazaphosphorine; P450; Cyclophosphamide; Ifosfamide Download date 28/09/2021 14:57:20 Link to Item http://hdl.handle.net/10713/2779 Curriculum Vitae Duan Wang Department of Pharmaceutical Sciences, School of Pharmacy, University of Maryland, Baltimore. 20 Penn Street, Baltimore, MD 21201,USA Phone: (410) 706 2123 E-mail: [email protected] Degree and Date to be Conferred: Ph.D., 2013 EDUCATION PhD candidate in Pharmaceutical Sciences Sep 2007~Apr 2013 University of Maryland School of Pharmacy, Maryland, USA Bachelor of Science Sep2003~Jun 2007 Huazhong University of Science & Technology, Wuhan, China ACADEMIC EXPERIENCE Research Assistant Aug 2007~Apr 2013 University of Maryland School of Pharmacy, Maryland, USA • Established an innovative cell-based co-culture system to quantitatively monitor the metabolism of cyclophosphamide and its main active metabolite, 4- hydroxycyclophosphamide using LC-MS/MS. Tested this assay on mouse, rat and human primary hepatocytes. Monitored the formation of metabolite in a time- and dose-dependent manner with quantification. • Established a 96-well plate based reporter assay and screened a 800-herbal medicine bank, identified several compounds of interest. • Explored the role of Constitutive Androstane Receptor (CAR) in the induction of Drug Metabolizing Enzymes by a variety of drugs, drug candidates and other compounds. -

Insights Into Immunogenic Cell Death in Onco-Therapies
cancers Review Restoring the Immunity in the Tumor Microenvironment: Insights into Immunogenic Cell Death in Onco-Therapies Ángela-Patricia Hernández 1 , Pablo Juanes-Velasco 1 , Alicia Landeira-Viñuela 1, Halin Bareke 1,2, Enrique Montalvillo 1, Rafael Góngora 1 and Manuel Fuentes 1,3,* 1 Department of Medicine and General Cytometry Service-Nucleus, CIBERONC CB16/12/00400, Cancer Research Centre (IBMCC/CSIC/USAL/IBSAL), 37007 Salamanca, Spain; [email protected] (Á.-P.H.); [email protected] (P.J.-V.); [email protected] (A.L.-V.); [email protected] (H.B.); [email protected] (E.M.); [email protected] (R.G.) 2 Department of Pharmaceutical Biotechnology, Faculty of Pharmacy, Institute of Health Sciences, Marmara University, 34722 Istanbul, Turkey 3 Proteomics Unit, Cancer Research Centre (IBMCC/CSIC/USAL/IBSAL), 37007 Salamanca, Spain * Correspondence: [email protected]; Tel.: +34-923-294-811 Simple Summary: Since the role of immune evasion was included as a hallmark in cancer, the idea of cancer as a single cell mass that replicate unlimitedly in isolation was dissolved. In this sense, cancer and tumorigenesis cannot be understood without taking into account the tumor microenvironment (TME) that plays a crucial role in drug resistance. Immune characteristics of TME can determine the success in treatment at the same time that antitumor therapies can reshape the immunity in TME. Here, we collect a variety of onco-therapies that have been demonstrated to induce an interesting Citation: Hernández, Á.-P.; immune response accompanying its pharmacological action that is named as “immunogenic cell Juanes-Velasco, P.; Landeira-Viñuela, death”. As this report shows, immunogenic cell death has been gaining importance in antitumor A.; Bareke, H.; Montalvillo, E.; therapy and should be studied in depth as well as taking into account other applications that may Góngora, R.; Fuentes, M. -

DOT1L Inhibitor EPZ-5676 Displays Synergistic Antiproliferative
JPET Fast Forward. Published on July 3, 2014 as DOI: 10.1124/jpet.114.214577 JPETThis Fast article Forward. has not been Published copyedited and on formatted. July 3, The 2014 final as version DOI:10.1124/jpet.114.214577 may differ from this version. JPET#214577 DOT1L Inhibitor EPZ-5676 Displays Synergistic Antiproliferative Activity in Combination with Standard of Care Drugs and Hypomethylating Agents in MLL-Rearranged Leukemia Cells Christine R. Klaus, Dorothy Iwanowicz, L. Danielle Johnston, Carly A. Campbell, Jesse J.Smith, Mikel P. Moyer, Robert A. Copeland, Edward J. Olhava, Margaret Porter Scott, Roy M. Pollock, Scott R. Daigle, and Alejandra Raimondi Downloaded from Author Affiliations: Epizyme Inc., 400 Technology Square, 4th floor, Cambridge MA 02139, USA: CRK, DI, LDJ, CAC, JJS, MPM, RAC, EJO, MPS, RMP, SRD, AR jpet.aspetjournals.org at ASPET Journals on September 24, 2021 1 Copyright 2014 by the American Society for Pharmacology and Experimental Therapeutics. JPET Fast Forward. Published on July 3, 2014 as DOI: 10.1124/jpet.114.214577 This article has not been copyedited and formatted. The final version may differ from this version. JPET#214577 Running Title: Synergy of EPZ-5676 with AML SOC in MLL-r Leukemia Corresponding author: Alejandra Raimondi, 400 Technology Square, 4th floor, Cambridge, MA 02139 Phone: +1 (617) 500-0708 Fax: +1 (617) 349-0707 Email: [email protected] Number of text pages: 45 Downloaded from Number of figures: 6 Number of tables: 1 jpet.aspetjournals.org Number of words in the Abstract: 255 Number of words in the Introduction: 711 at ASPET Journals on September 24, 2021 Number of words in the Discussion: 1293 Number of References: 47 List of nonstandard abbreviations: ALL Acute Lymphoblastic Leukemia AML Acute Myeloid Leukemia BSA Bovine Serum Albumin CI Combination index DMSO Dimethyl sulfoxide DNMT DNA methyltransferase Fa Fractional effect 2 JPET Fast Forward. -

Recent Advances in Myelodysplastic
Haematologica 1998; 83:910-935 decision making and problem solving Recent Advances in Myelodysplastic Syndromes Guest Editors: Miguel Angel Sanz, Guillermo Sanz & Teresa Vallespí A patient-oriented approach to treatment of myelodysplastic syndromes MARIO CAZZOLA,* JEANNE E. ANDERSON,° ARNOLD GANSER,# EVA HELLSTRÖM-LINDBERG@ *Department of Internal Medicine and Medical Therapy, Section of Internal Medicine and Medical Oncology, University of Pavia School of Medicine, IRCCS Policlinico S. Matteo, Pavia, Italy; °Department of Medicine, Division of Hematology, University of Texas Health Science Center at San Antonio, USA; #Department of Hematology/Oncology, Hannover Univer- sity Medical School, Hannover, Germany; @Department of Hematology, Huddinge University Hospital, Huddinge, Sweden Abstract Background and Objective. There are several thera- experience an improvement in quality of life but very peutic options for patients with myelodysplastic syn- few studies have addressed this question so far. The drome (MDS) but most of them are poorly effective majority of MDS patients still rely upon supportive and the potentially curative ones are available only for therapy. A clinical decision path based on findings of a minority of individuals. The aim of this article is to clinical trials and the patient’s expectations can help define a rational basis for a patient-oriented approach physicians in decision making. Because of the inade- to treatment of MDS. quacies of all current treatment modalities, participi- ation in clinical trials should always be encouraged. Evidence and Information Sources. All four authors ©1998, Ferrata Storti Foundation have done clinical studies of treatment of MDS, including stem cell transplantation, intensive and low- dose chemotherapy, and use of hematopoietic growth Key words: myelodysplastic syndromes, therapy factors. -

Stembook 2018.Pdf
The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 WHO/EMP/RHT/TSN/2018.1 © World Health Organization 2018 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances. Geneva: World Health Organization; 2018 (WHO/EMP/RHT/TSN/2018.1). Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data.