Applications of Resonance Raman Spectroscopy to the Study of Bioinorganic Macromolecules
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

DEVELOPMENT of a RAMAN SPECTROMETER to STUDY SURFACE-ENHANCED RAMAN SCATTERING by Nandita Biswas, Ridhima Chadha, Sudhir Kapoor, Sisir K
BARC/2011/E/003 BARC/2011/E/003 DEVELOPMENT OF A RAMAN SPECTROMETER TO STUDY SURFACE-ENHANCED RAMAN SCATTERING by Nandita Biswas, Ridhima Chadha, Sudhir Kapoor, Sisir K. Sarkar and Tulsi Mukherjee Radiation & Photochemistry Division 2011 BARC/2011/E/003 GOVERNMENT OF INDIA ATOMIC ENERGY COMMISSION BARC/2011/E/003 DEVELOPMENT OF A RAMAN SPECTROMETER TO STUDY SURFACE-ENHANCED RAMAN SCATTERING by Nandita Biswas, Ridhima Chadha, Sudhir Kapoor, Sisir K. Sarkar and Tulsi Mukherjee Radiation & Photochemistry Division BHABHA ATOMIC RESEARCH CENTRE MUMBAI, INDIA 2011 BARC/2011/E/003 BIBLIOGRAPHIC DESCRIPTION SHEET FOR TECHNICAL REPORT (as per IS : 9400 - 1980) 01 Security classification : Unclassified 02 Distribution : External 03 Report status : New 04 Series : BARC External 05 Report type : Technical Report 06 Report No. : BARC/2011/E/003 07 Part No. or Volume No. : 08 Contract No. : 10 Title and subtitle : Development of a Raman spectrometer to study surface-enhanced Raman scattering 11 Collation : 31 p., 15 figs. 13 Project No. : 20 Personal author(s) : Nandita Biswas; Ridhima Chadha; Sudhir Kapoor; Sisir K. Sarkar; Tulsi Mukherjee 21 Affiliation of author(s) : Radiation and Photochemistry Division , Bhabha Atomic Research Centre, Mumbai 22 Corporate author(s) : Bhabha Atomic Research Centre, Mumbai - 400 085 23 Originating unit : Radiation and Photochemistry Division, BARC, Mumbai 24 Sponsor(s) Name : Department of Atomic Energy Type : Government Contd... BARC/2011/E/003 30 Date of submission : January 2011 31 Publication/Issue date : February 2011 40 Publisher/Distributor : Head, Scientific Information Resource Division, Bhabha Atomic Research Centre, Mumbai 42 Form of distribution : Hard copy 50 Language of text : English 51 Language of summary : English, Hindi 52 No. -
The Term Fluorescence Was Coined by Stokes Circa 1850 to Name A
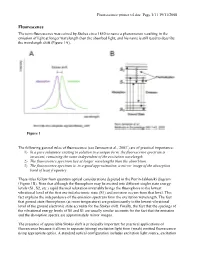
Fluorescence primer v4.doc Page 1/11 19/11/2008 Fluorescence The term fluorescence was coined by Stokes circa 1850 to name a phenomenon resulting in the emission of light at longer wavelength than the absorbed light, and his name is still used to describe the wavelength shift (Figure 1A). Figure 1 The following general rules of fluorescence (see Jameson et al., 20031) are of practical importance: 1) In a pure substance existing in solution in a unique form, the fluorescence spectrum is invariant, remaining the same independent of the excitation wavelength. 2) The fluorescence spectrum lies at longer wavelengths than the absorbtion. 3) The fluorescence spectrum is, to a good approximation, a mirror image of the absorption band of least frequency These rules follow from quantum optical considerations depicted in the Perrin-Jablonski diagram (Figure 1B). Note that although the fluorophore may be excited into different singlet state energy levels (S1, S2, etc.) rapid thermal relaxation invariably brings the fluorophore to the lowest vibrational level of the first excited electronic state (S1) and emission occurs from that level. This fact explains the independence of the emission spectrum from the excitation wavelength. The fact that ground state fluorophores (at room temperature) are predominantly in the lowest vibrational level of the ground electronic state accounts for the Stokes shift. Finally, the fact that the spacings of the vibrational energy levels of S0 and S1 are usually similar accounts for the fact that the emission and the absorption spectra are approximately mirror images. The presence of appreciable Stokes shift is principally important for practical applications of fluorescence because it allows to separate (strong) excitation light from (weak) emitted fluorescence using appropriate optics. -

Resonant Raman Spectroscopy of Nanotubes
10.1098/rsta.2004.1444 Resonant Raman spectroscopy of nanotubes By Christian Thomsen1, Stephanie Reich2 and Janina Maultzsch1 1Technische Universit¨at Berlin, Hardenbergstraße 36, 10623 Berlin, Germany 2Department of Engineering, University of Cambridge, Trumpington Street, Cambridge CB2 1PZ, UK ([email protected]) Published online 28 September 2004 Single and double resonances in Raman scattering are introduced and six criteria for the observation and identification of double resonances stated. The experimental situation in carbon nanotubes is reviewed in view of these criteria. The evidence for the D mode and the high-energy mode is found to be overwhelming for a double- resonance process to take place, whereas the nature of the radial breathing-mode Raman process remains undecided at this point. Consequences for the application of Raman scattering to the characterization of nanotubes are discussed. Keywords: carbon nanotubes; double resonance; Raman scattering; defects 1. Introduction Raman scattering in carbon nanotubes has developed into a method of choice in the investigation of their physical properties and their characterization. The study of electronic resonances in the Raman spectra, a method which has been used exten- sively, for example, in work on semiconductors (Cardona 1982), gives us a wealth of information about the electronic band structure of a material. This is also true for the Raman work on carbon nanotubes, where resonance studies have moved into the focus of research. Traditional Raman studies of carbon nanotubes focus on the radial breathing mode (RBM), a mode where all atoms vibrate in phase in the radial direction (Dresselhaus et al. 1995). Its frequency, as can easily be shown (Jishi et al. -

Raman Scattering and Fluorescence
Fluorescence 01 Raman Scattering and Fluorescence Introduction The existence of such virtual states also explains why the non-resonance Raman effect Raman scattering and Fluorescence emission does not depend on the wavelength of the are two competing phenomena, which have excitation, since no real states are involved in similar origins. Generally, a laser photon this interaction mechanism. In fact, the Raman bounces off a molecule and looses a certain spectrum generally does not depend on the amount of energy that allows the molecule to laser excitation. vibrate (Stokes process). The scattered photon is therefore less energetic and the associated However, when the energy of the excitation light exhibits a frequency shift. The various photon gets close to the transition energy frequency shifts associated with different between two electronic states, one then deals molecular vibrations give rise to a spectrum, with resonance Raman or resonance that is characteristic of a specific compound. fluorescence (fig.1, case (d)). The basic difference between these two processes is In contrast, fluorescence or luminescence related to the time scales involved, as well as emission follows an absorption process. For a with the nature of the so-called intermediate better understanding, one can refer to the states. In contrast with resonant fluorescent, diagram below. relaxed fluorescence results from the emission of a photon from the lowest vibrational level of an excited electronic state, following a direct absorption of the photon and relaxation of the molecule from its vibrationally excited level of the electronic state back to the lowest vibrational level of the electronic state. A fluorescence process typically requires more than 10-9 s. -

The Electromagnetic Spectrum
The Electromagnetic Spectrum Wavelength/frequency/energy MAP TAP 2003-2004 The Electromagnetic Spectrum 1 Teacher Page • Content: Physical Science—The Electromagnetic Spectrum • Grade Level: High School • Creator: Dorothy Walk • Curriculum Objectives: SC 1; Intro Phys/Chem IV.A (waves) MAP TAP 2003-2004 The Electromagnetic Spectrum 2 MAP TAP 2003-2004 The Electromagnetic Spectrum 3 What is it? • The electromagnetic spectrum is the complete spectrum or continuum of light including radio waves, infrared, visible light, ultraviolet light, X- rays and gamma rays • An electromagnetic wave consists of electric and magnetic fields which vibrates thus making waves. MAP TAP 2003-2004 The Electromagnetic Spectrum 4 Waves • Properties of waves include speed, frequency and wavelength • Speed (s), frequency (f) and wavelength (l) are related in the formula l x f = s • All light travels at a speed of 3 s 108 m/s in a vacuum MAP TAP 2003-2004 The Electromagnetic Spectrum 5 Wavelength, Frequency and Energy • Since all light travels at the same speed, wavelength and frequency have an indirect relationship. • Light with a short wavelength will have a high frequency and light with a long wavelength will have a low frequency. • Light with short wavelengths has high energy and long wavelength has low energy MAP TAP 2003-2004 The Electromagnetic Spectrum 6 MAP TAP 2003-2004 The Electromagnetic Spectrum 7 Radio waves • Low energy waves with long wavelengths • Includes FM, AM, radar and TV waves • Wavelengths of 10-1m and longer • Low frequency • Used in many -

Technical Application Note
Compact Rugged Spectrometers - A Universe of Spectroscopy Systems Technical Application Note Vibrational Spectroscopy: Infrared vs. Raman StellarNet, Inc. Tampa, Fl USA Tony Rizzuto, PhD April 12, 2017 Vibrational spectroscopies are integral in analyzing some of the most fundamentally important processes in physical chemistry: molecular vibrations. While there are many different experimental techniques used to analyze those vibrations, most are variations of the “Big Two,” FTIR and Raman scattering spectroscopies. This article aims to analyze the benefits and drawbacks of each of the “Big Two,” with the hope that it helps one choose the correct technique for one’s research interests. 1. What are the “Big Two?” Fourier Transform Infrared Spectroscopy Raman spectroscopy relies on inelastic (FTIR) is a simple absorption measurement scattering phenomenon that probes the where the detector measures the molecular vibration. Where FTIR will use a absorbance of infrared radiation by the broadband IR source, Raman spectroscopy sample. Each sample will absorb different typically uses a narrow-band, amounts of each frequency resulting in a monochromatic light source in order to “chemical fingerprint” that is the FTIR excite the vibrations of the molecule in your spectrum. sample. 2. Selection Rules: What molecular vibrations are being probed? Imagining a molecular bond vibration with the traditional ball and spring model (Figure 1a) and its resultant harmonic oscillator depiction (Figure 1b) allows us to calculate the selection rules for vibrational transitions to be = 1. However, that does not tell the whole story. In order to truly differentiate FTIR from Raman Spectroscopy, we must think about it on a molecular level. Compact Rugged Spectrometers - A Universe of Spectroscopy Systems Technical Application Note A B Figure 1: A. -

Including Far Red in an LED Lighting Spectrum
technically speaking BY ERIK RUNKLE Including Far Red in an LED Lighting Spectrum Far red (FR) is a one of the radiation (or light) wavebands larger leaves can be desired for other crops. that regulates plant growth and development. Many people We have learned that blue light (and to a smaller extent, consider FR as radiation with wavelengths between 700 and total light intensity) can influence the effects of FR. When the 800 nm, although 700 to 750 nm is, by far, the most active. intensity of blue light is high, adding FR only slightly increases By definition, FR is just outside the photosynthetically active extension growth. Therefore, the utility of including FR in an radiation (PAR) waveband, but it can directly and indirectly indoor lighting spectrum is greater under lower intensities increase growth. In addition, it can accelerate of blue light. One compelling reason to deliver at least some flowering of some crops, especially long-day plants, FR light indoors is to induce early flowering of young plants, which are those that flower when the nights are short. especially long-day plants. As we learn more about the effects of FR on plants, growers sometimes wonder, is it beneficial to include FR in a light-emitting diode (LED) spectrum? "As the DLI increases, Not surprisingly, the answer is, it depends on the application and crop. In the May 2016 issue of GPN, I wrote about the the utility of FR in effects of FR on plant growth and flowering (https:// bit.ly/2YkxHCO). Briefly, leaf size and stem length photoperiodic lighting increase as the intensity of FR increases, although the magnitude depends on the crop and other characteristics of the light environment. -

Stokes Shift
Stokes shift ticular molecular structure. If a material has a direct bandgap in the range of visible light, the light shining on it is absorbed, causing electrons to become excited to a higher energy state. The electrons remain in the ex- cited state for about 10−8 seconds. This number varies over several orders of magnitude depending on the sam- ple, and is known as the fluorescence lifetime of the sam- ple. After losing a small amount of energy in some way (hence the longer wavelength), the molecule returns to the ground state and energy is emitted. 2 References Absorption and emission spectra of Rhodamine 6G with ~25 nm [1] Gispert, J.R. (2008). Coordination Chemistry. Wiley- Stokes shift VCH. p. 483. ISBN 3-527-31802-X. [2] Albani, J.R. (2004). Structure and Dynamics of Macro- Not to be confused with Stark shift. molecules: Absorption and Fluorescence Studies. Elsevier. p. 58. ISBN 0-444-51449-X. Stokes shift is the difference (in wavelength or frequency [3] Lakowicz, J.R. 1983. Principles of Fluorescence Spec- units) between positions of the band maxima of the troscopy, Plenum Press, New York. ISBN 0-387-31278- absorption and emission spectra (fluorescence and Raman 1. being two examples) of the same electronic transition.[1] [4] Guilbault, G.G. 1990. Practical Fluorescence, Second It is named after Irish physicist George G. Stokes.[2][3][4] Edition, Marcel Dekker, Inc., New York. ISBN 0-8247- When a system (be it a molecule or atom) absorbs a 8350-6. photon, it gains energy and enters an excited state. -

Electromagnetic Spectrum
Electromagnetic Spectrum Why do some things have colors? What makes color? Why do fast food restaurants use red lights to keep food warm? Why don’t they use green or blue light? Why do X-rays pass through the body and let us see through the body? What has the radio to do with radiation? What are the night vision devices that the army uses in night time fighting? To find the answers to these questions we have to examine the electromagnetic spectrum. FASTER THAN A SPEEDING BULLET MORE POWERFUL THAN A LOCOMOTIVE These words were used to introduce a fictional superhero named Superman. These same words can be used to help describe Electromagnetic Radiation. Electromagnetic Radiation is like a two member team racing together at incredible speeds across the vast regions of space or flying from the clutches of a tiny atom. They travel together in packages called photons. Moving along as a wave with frequency and wavelength they travel at the velocity of 186,000 miles per second (300,000,000 meters per second) in a vacuum. The photons are so tiny they cannot be seen even with powerful microscopes. If the photon encounters any charged particles along its journey it pushes and pulls them at the same frequency that the wave had when it started. The waves can circle the earth more than seven times in one second! If the waves are arranged in order of their wavelength and frequency the waves form the Electromagnetic Spectrum. They are described as electromagnetic because they are both electric and magnetic in nature. -

Resonance Enhancement of Raman Spectroscopy: Friend Or Foe?
www.spectroscopyonline.com ® Electronically reprinted from June 2013 Volume 28 Number 6 Molecular Spectroscopy Workbench Resonance Enhancement of Raman Spectroscopy: Friend or Foe? The presence of electronic transitions in the visible part of the spectrum can provide enor- mous enhancement of the Raman signals, if these electronic states are not luminescent. In some cases, the signals can increase by as much as six orders of magnitude. How much of an enhancement is possible depends on several factors, such as the width of the excited state, the proximity of the laser to that state, and the enhancement mechanism. The good part of this phenomenon is the increased sensitivity, but the downside is the nonlinearity of the signal, making it difficult to exploit for analytical purposes. Several systems exhibiting enhancement, such as carotenoids and hemeproteins, are discussed here. Fran Adar he physical basis for the Raman effect is the vibra- bound will be more easily modulated. So, because tional modulation of the electronic polarizability. electrons are more loosely bound than electrons, the T In a given molecule, the electronic distribution is polarizability of any unsaturated chemical functional determined by the atoms of the molecule and the electrons group will be larger than that of a chemically saturated that bind them together. When the molecule is exposed to group. Figure 1 shows the spectra of stearic acid (18:0) and electromagnetic radiation in the visible part of the spec- oleic acid (18:1). These two free fatty acids are both con- trum (in our case, the laser photons), its electronic dis- structed from a chain of 18 carbon atoms, in one case fully tribution will respond to the electric field of the photons. -

Fiber Amplifiers and Fiber Lasers Based on Stimulated Raman
micromachines Review Fiber Amplifiers and Fiber Lasers Based on Stimulated Raman Scattering: A Review Luigi Sirleto * and Maria Antonietta Ferrara National Research Council (CNR), Institute of Applied Sciences and Intelligent Systems, Via Pietro Castellino 111, 80131 Naples, Italy; [email protected] * Correspondence: [email protected] Received: 10 January 2020; Accepted: 24 February 2020; Published: 26 February 2020 Abstract: Nowadays, in fiber optic communications the growing demand in terms of transmission capacity has been fulfilling the entire spectral band of the erbium-doped fiber amplifiers (EDFAs). This dramatic increase in bandwidth rules out the use of EDFAs, leaving fiber Raman amplifiers (FRAs) as the key devices for future amplification requirements. On the other hand, in the field of high-power fiber lasers, a very attractive option is provided by fiber Raman lasers (FRLs), due to their high output power, high efficiency and broad gain bandwidth, covering almost the entire near-infrared region. This paper reviews the challenges, achievements and perspectives of both fiber Raman amplifier and fiber Raman laser. They are enabling technologies for implementation of high-capacity optical communication systems and for the realization of high power fiber lasers, respectively. Keywords: stimulated raman scattering; fiber optics; amplifiers; lasers; optical communication systems 1. Introduction Optical communication systems require optoelectronic devices, such as sources, detectors and so on, and utilize fiber optics to transmit the light carrying the signals impressed by modulators. Optical fibers are affected by chromatic dispersion, losses, and nonlinearity. Dispersion control is, usually, achieved via fiber geometry and material composition. Losses limit the transmission distance in modern long haul fiber-optic communication systems, so in order to boost a weak signal, optical amplifiers have been developed. -

Article Intends to Provide a for the Necessary Virtual Electronic Brief Overview of the Differences and Transition
ADVANCES IN RAMAN TECHNIQUES Laser requirements and advances for Raman techniques Andreas Isemann Laser Quantum GmbH, 78467 Konstanz, Germany INTRODUCTION 473 nm and 1064 nm, a narrow Raman scattering as a probe of bandwidth output of few tens of GHz vibrational transitions has made or below 1 MHz if needed within the leaps and bounds since its discovery, linewidth of vibrational transitions and various schemes based on this for high resolution, low noise (less phenomenon have been developed than 0.02%) and excellent beam with great success. quality (fundamental transversal Applications range from basic electromagnetic mode TEM00) scientific research, to medical and provides optimised performance industrial instrumentation. Some for the resolution of the Raman schemes utilise linear Raman measurement needed. scattering, whilst others take advantage The wavelength is chosen based of high peak-power fields to probe on the sample under investigation, nonlinear Raman responses. with 532 nm being commonly used This article intends to provide a for the necessary virtual electronic brief overview of the differences and transition. In the following section, benefits, together with the laser source four examples from different areas of requirements and the advancements Raman applications show the diverse in techniques enabled by recent applications of linear Raman and what developments in lasers. advances have been achieved. An example of studying a real-world LINEAR RAMAN application, the successful control of Figure 1 An example of the RR microfluidic device counting of The advent of the laser in providing a food quality using Raman spectroscopy photosynthetic microorganisms. As the cells of the model strain high-intensity coherent light source and multivariate analysis, is described Synechocystis sp.