Inv Dup (22), Del (22)(Q11) and R (22) in the Father of a Child With
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Molecular Delineation of Small Supernumerary Marker
Zhou et al. Molecular Cytogenetics (2020) 13:19 https://doi.org/10.1186/s13039-020-00486-2 RESEARCH Open Access Molecular delineation of small supernumerary marker chromosomes using a single nucleotide polymorphism array Lili Zhou1, Zhaoke Zheng1, Lianpeng Wu2, Chenyang Xu1, Hao Wu1, Xueqin Xu1 and Shaohua Tang1,2* Abstract Background: Defining the phenotype-genotype correlation of small supernumerary marker chromosomes (sSMCs) remains a challenge in prenatal diagnosis. We karyotyped 20,481 amniotic fluid samples from pregnant women and explored the molecular characteristics of sSMCs using a single nucleotide polymorphism (SNP) array. Results: Out of the 20,481 samples, 15 abnormal karyotypes with sSMC were detected (frequency: 0.073%) and the chromosomal origin was successfully identified by SNP array in 14 of them. The origin of sSMCs were mainly acrocentric-derived chromosomes and the Y chromosome. Two cases of sSMC combined with uniparental disomy (UPD) were detected, UPD(1) and UPD(22). More than half of the cases of sSMC involved mosaicism (8/15) and pathogenicity (9/15) in prenatal diagnosis. A higher prevalence of mosaicism for non-acrocentric chromosomes than acrocentric chromosomes was also revealed. One sSMC derived from chromosome 3 with a neocentromere revealed a 24.99-Mb pathogenic gain of the 3q26.31q29 region on the SNP array, which presented as an abnormal ultrasound indicating nasal bone hypoplasia. Conclusion: The clinical phenotypes of sSMCs are variable and so further genetic testing and parental karyotype analysis are needed to confirm the characteristics of sSMCs. The SNP array used here allows a detailed characterisation of the sSMC and establishes a stronger genotype-phenotype correlation, thus allowing detailed genetic counselling for prenatal diagnosis. -

General Contribution
24 Abstracts of 37th Annual Meeting A1 A SCREENING METHOD FOR FRAGILE X MUTATION: DETECTION OF THE CGG REPEAT IN FMR-1 GENE BY PCR WITH BIOTIN-LABELED PRIMER. ..Eiji NANBA, Kousaku OHNO and Kenzo TAKESHITA Division of Child Neurology, Institute of Neurological Sciences, Tot- tori University School of Medicine. Yonago We have developed a new polymerase chain reaction(PCR)-based method for detection of the CGG repeat in FMR-1 gene. No specific product from PCR was detected on the gel with ethidium bromide staining, because 7-deaza-2'-dGTP is necessary for amplification of this repeat. Biotin-labeled primer was used for PCR and the product was transferred to a nylon membrane followed the detection of biotin by Smilight kit. The size of PCR product from normal control were slightly various and around 300bp. No PCR product was detected from 3 fragile X male patients in 2 families diagnosed by cytogenetic examination. This method is useful for genetic screen- ing of male mental retardation patients to exclude the fragile X mutation. A2 DNA ANALYSISFOR FRAGILE X SYNDROME Osamu KOSUDA,Utak00GASA, ~.ideynki INH, a~ji K/NAGIJCltI, and Kazumasa ]tIKIJI (SILL Inc., Tokyo) Fragile X syndrome is X-linked disease having the amplification of (CG6)n repeat sequence in the chromsomeXq27.3. We performed Southern blot analysis using three probes recognized repetitive sequence resion. Normal controle showed 5.2Kb with Eco RI digest and 2.7Kb with Eco RI/Bss ttII digest as the germ tines by the Southern blot analysis. However, three cell lines established fro~ unrelated the patients with fragile X showed the abnormal bands between 5.2 and 7.7Kb with Eco RI digest, and between 2.7 and 7.7Kb with Eco aI/Bss HII digest. -

Soonerstart Automatic Qualifying Syndromes and Conditions
SoonerStart Automatic Qualifying Syndromes and Conditions - Appendix O Abetalipoproteinemia Acanthocytosis (see Abetalipoproteinemia) Accutane, Fetal Effects of (see Fetal Retinoid Syndrome) Acidemia, 2-Oxoglutaric Acidemia, Glutaric I Acidemia, Isovaleric Acidemia, Methylmalonic Acidemia, Propionic Aciduria, 3-Methylglutaconic Type II Aciduria, Argininosuccinic Acoustic-Cervico-Oculo Syndrome (see Cervico-Oculo-Acoustic Syndrome) Acrocephalopolysyndactyly Type II Acrocephalosyndactyly Type I Acrodysostosis Acrofacial Dysostosis, Nager Type Adams-Oliver Syndrome (see Limb and Scalp Defects, Adams-Oliver Type) Adrenoleukodystrophy, Neonatal (see Cerebro-Hepato-Renal Syndrome) Aglossia Congenita (see Hypoglossia-Hypodactylia) Aicardi Syndrome AIDS Infection (see Fetal Acquired Immune Deficiency Syndrome) Alaninuria (see Pyruvate Dehydrogenase Deficiency) Albers-Schonberg Disease (see Osteopetrosis, Malignant Recessive) Albinism, Ocular (includes Autosomal Recessive Type) Albinism, Oculocutaneous, Brown Type (Type IV) Albinism, Oculocutaneous, Tyrosinase Negative (Type IA) Albinism, Oculocutaneous, Tyrosinase Positive (Type II) Albinism, Oculocutaneous, Yellow Mutant (Type IB) Albinism-Black Locks-Deafness Albright Hereditary Osteodystrophy (see Parathyroid Hormone Resistance) Alexander Disease Alopecia - Mental Retardation Alpers Disease Alpha 1,4 - Glucosidase Deficiency (see Glycogenosis, Type IIA) Alpha-L-Fucosidase Deficiency (see Fucosidosis) Alport Syndrome (see Nephritis-Deafness, Hereditary Type) Amaurosis (see Blindness) Amaurosis -

22Q12 and 22Q13 Duplications
22q12 and 22q13 duplications rarechromo.org Duplications of 22q12 and 22q13 A duplication of 22q12 and/or 22q13 is a very rare genetic condition in which the cells of the body have a small but variable amount of extra genetic material from one of the body’s 46 chromosomes – chromosome 22. For healthy development, chromosomes should contain just the right amount of genetic material (DNA) – not too much and not too little. Like most other chromosome disorders, having an extra part of chromosome 22 may increase the risk of birth defects, developmental delay and intellectual disability. However, there is individual variation. Background on Chromosomes Chromosomes are structures which contain our DNA and are found in almost every cell of the body. Every chromosome contains thousands of genes which may be thought of as individual instruction booklets (or recipes) that contain all the genetic information telling the body how to develop, grow and function. Chromosomes (and genes) usually come in pairs with one member of each chromosome pair being inherited from each parent. Most cells of the human body have a total of 46 (23 pairs of) chromosomes. The egg and the sperm cells, however have 23 unpaired chromosomes, so that when the egg and sperm join together at conception, the chromosomes pair up and the number is restored to 46. Of these 46 chromosomes, two are the sex chromosomes that determine gender. Females have two X chromosomes and males have one X chromosome and one Y chromosome. The remaining 44 chromosomes are grouped in 22 pairs, numbered 1 to 22 approximately from the largest to the smallest. -

Case Report: Description of a Patient with Trisomy 22 in Inbred Line
December 2013, Vol. 7, No. 12, pp. 1312-1316 Journal of Life Sciences, ISSN 1934-7391, USA D DAVID PUBLISHING Case Report: Description of a Patient with Trisomy 22 in Inbred Line Leandro Gutiérrez1, Flavia Leveroni1, Cristina Mayer2, Jorge Doldán2, Amada Rolón1, Ana Melnichuk1, Alejandro Laudicina3, Sonia Bageston2 and Alberto Fenocchio1 1. Laboratory of Human Genetics and Cytogenetic, School of Natural, Chemistry and Exact Sciences, National University of Misiones, Posadas, Misiones N3300MLL, Argentina 2. Laboratory of Cytogenetic, Dr. Ramón Madariaga Hospital, Posadas, Misiones N3300MLL, Argentina 3. Lexel SRL, División In Vitro, Buenos Aires City, Buenos Aires C1135ABO, Argentina Received: September 25, 2013 / Accepted: November 28, 2013 / Published: December 30, 2013. Abstract: Long arm trisomy of chromosome 22 or cat eye syndrome (OMIM#115470) is a disease with an enormous variability of clinical features, ranging from minor malformations like hypertelorism, to major ones, as congenital heart and renal disorders, combined with variable growth retardation. The authors report a case of a newborn female with clinical features of cat eye syndrome with trisomy 22 in inbred line, who died 35 days after birth. The clinical features at the time of diagnosis were: left preauricular appendix, low-set ears, hypertelorism, mongoloid palpebral apertures, right-sided microphthalmia, left-sided anophthalmia, cleft lip and palate, short neck, anomalous pulmonary venous return, severe lung hypertension, hyperechogenic little kidneys and clinodactyly of the fifth finger on the left side. Cerebral ultrasound showed dilatation of both lateral ventricles, with a callosum corpus difficult to evaluate. The cytogenetics diagnostic was made from peripheral blood by conventional cytogenetics techniques in two different laboratories, and confirmed by fluorescent in situ hybridization. -
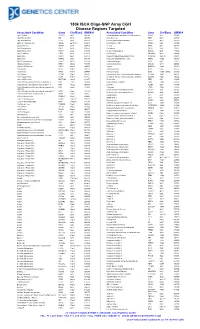
180K ISCA Oligo-SNP Array CGH Disease Regions Targeted
180k ISCA Oligo-SNP Array CGH Disease Regions Targeted Associated Condition Gene Chr/Band OMIM # Associated Condition Gene Chr/Band OMIM # 1p31 Deletion DIRAS3 1p31 605193 Chondrodysplasia punctata, X-linked recessive CDPX1 Xp22 302950 1p36 Microdeletion SKI 1p36 607872 Choroideremia CHM Xp21 303100 1p36 Microdeletion TP73 1p36 607872 Chronic granulomatous disease CYBB Xp11 306400 2p15-16.1 Microdeletion Multiple 2p15-16.1 612513 Chronic pancreatitis SPINK1 5q32 167800 2q37.3 Deletion HDAC4 2q37 600430 Cleft lip MSX1 4p16 608874 3q29 Microdeletion PAK2 3q29 609425 Cleft palate SATB2 2q32 119540 6p24 Microdeletion FKHL7 6p24 612852 Cleidocranial dysplasia RUNX2 6p21 119600 8p23.1 Deletion CTSB 8p23 116810 Coffin-Lowry RPS6KA3 Xp22 303600 9p Deletion DMRT1 9p24 158170 Congenital adrenal hyperplasia (CAH) CYP21A2 6p21.32 201910 9p Deletion DMRT2 9p24 158170 Congenital diaphragmatic hernia NR2F2 15q26 142340 9q34.3 Microdeletion EHMT1 9q34.3 610253 Cornelia de Lange 1 NIPBL 5p13 122470 10q22-23 Deletion GRID1 10q22 610659 Cornelia de Lange 2 SMC1L1 Xp11 300590 12q14.1-q15 Deletion GRIP1 12q14 604597 Cowden BMPR1A 10q23 158350 13q Deletion GPC5 13q31 602446 Craniosynostosis MSX2 5q35 604757 13q Deletion GPC6 13q31 604404 Craniosynostosis SOX6 11p15.1-p15.2 218350 13q Deletion PCDH9 13q21 603581 Creatine deficiency / X-linked mental retardation SLC6A8 16p11 300352 14q11-q22 Deletion CHD8 14q11 613457 Creatine deficiency / X-linked mental retardation SLC6A8 16p11 300352 14q11-q22 Deletion SUPT16H 14q11 613457 Cri-du-Chat TERT 5p15 123450 14q22 -

Aarskog Syndrome Parents Support Group
Aarskog Syndrome Parents Support Group http://www.familyvillage.wisc.edu/lib_aars.htm AboutFace USA (For people with facial differences) http://www.aboutfaceusa.org AboutFace International http://www.aboutfaceinternational.org ; http://aboutface.ca/ Abetalipoproteinemia (International) http://www.abetalipoproteinemia.org/ Accord Alliance (Disorders of Sexual Development) http://www.accordalliance.org/ Achalasia Support Groups http://achalasia.us/Support_Groups.html Acid Maltase Deficiency Association http://www.amda-pompe.org/ Acoustic Neuroma Association http://anausa.org/ Addison’s Disease Self Help Group UK http://www.addisons.org.uk/ Adinoid Cystic Carcinoma Organization International http://www.accoi.org/ Adinoid Cystic Carcinoma Research Foundation http://www.accrf.org/ Advocacy for Neuroacanthocytosis Patients http://www.naadvocacy.org/ AIS-DSD Support Group USA (Disorders of Sex Development) http://www.aisdsd.org Ais (Androgen Insensitivity Syndrome) Support Group http://www.aissg.org (UK based) AKU (Alkaptonuria) Society http://www.alkaptonuria.info/ Alagille Syndrome Alliance http://www.alagille.org/ ALS Association (Amyotrophic Lateral Sclerosis) http://www.alsa.org Alopecia Support Group (ASG) http://www.alopeciasupport.org/ Alpha-1 Association (Alpha-1 Antitrypsin Deficiency) http://www.alpha1.org/ Alpha-1 Canada http://www.alpha1canada.ca/ Alpha-1 Foundation http://www.alpha-1foundation.org/ Alport Syndrome Foundation http://www.alportsyndrome.org Alstrom Syndrome International http://www.alstrom.org/ Alternating Hemiplegia -

Chromosome 22
Chromosome 22 Description Humans normally have 46 chromosomes (23 pairs) in each cell. Two copies of chromosome 22, one copy inherited from each parent, form one of the pairs. Chromosome 22 is the second smallest human chromosome, spanning more than 51 million DNA building blocks (base pairs) and representing between 1.5 and 2 percent of the total DNA in cells. In 1999, researchers working on the Human Genome Project announced they had determined the sequence of base pairs that make up this chromosome. Chromosome 22 was the first human chromosome to be fully sequenced. Identifying genes on each chromosome is an active area of genetic research. Because researchers use different approaches to predict the number of genes on each chromosome, the estimated number of genes varies. Chromosome 22 likely contains 500 to 600 genes that provide instructions for making proteins. These proteins perform a variety of different roles in the body. Health Conditions Related to Chromosomal Changes The following chromosomal conditions are associated with changes in the structure or number of copies of chromosome 22. 22q11.2 deletion syndrome 22q11.2 deletion syndrome is a disorder involving heart defects, an opening in the roof of the mouth (a cleft palate), distinctive facial features, low calcium levels, and an increased risk of behavioral problems and mental illness such as schizophrenia. Most people with 22q11.2 deletion syndrome are missing about 3 million base pairs on one copy of chromosome 22 in each cell. The deletion occurs near the middle of the chromosome at a location designated as q11.2. This region contains 30 to 40 genes, but many of these genes have not been well characterized. -

An Autosomal Recessive Syndrome of Severe Mental Retardation, Cataract, Coloboma and Kyphosis Maps to the Pericentromeric Region of Chromosome 4
European Journal of Human Genetics (2009) 17, 125–128 & 2009 Macmillan Publishers Limited All rights reserved 1018-4813/09 $32.00 www.nature.com/ejhg SHORT REPORT An autosomal recessive syndrome of severe mental retardation, cataract, coloboma and kyphosis maps to the pericentromeric region of chromosome 4 Kimia Kahrizi1, Hossein Najmabadi1, Roxana Kariminejad1, Payman Jamali1, Mahdi Malekpour1, Masoud Garshasbi1,2, Hans Hilger Ropers2, Andreas Walter Kuss2 and Andreas Tzschach*,2 1Genetics Research Center, University of Social Welfare and Rehabilitation Sciences, Tehran, Iran; 2Department Human Molecular Genetics, Max Planck Institute for Molecular Genetics, Berlin, Germany We report on three siblings with a novel mental retardation (MR) syndrome who were born to distantly related Iranian parents. The clinical problems comprised severe MR, cataracts with onset in late adolescence, kyphosis, contractures of large joints, bulbous nose with broad nasal bridge, and thick lips. Two patients also had uni- or bilateral iris coloboma. Linkage analysis revealed a single 10.4 Mb interval of homozygosity with significant LOD score in the pericentromeric region of chromosome 4 flanked by SNPs rs728293 (4p12) and rs1105434 (4q12). This interval contains more than 40 genes, none of which has been implicated in MR so far. The identification of the causative gene defect for this syndrome will provide new insights into the development of the brain and the eye. European Journal of Human Genetics (2009) 17, 125–128; doi:10.1038/ejhg.2008.159; published online 10 September 2008 Keywords: mental retardation; autosomal recessive; consanguinity; cataract; coloboma; kyphosis Introduction Clinical report Mental retardation (MR) has a prevalence of about 2%,1 The pedigree of the family is shown in Figure 2a. -

Clinical Characteristics of a Sample of Patients with Cat Eye Et Al
ROSAArtigo RFM ET AL. Original CARACTERÍSTicAS CLÍNicAS DE UMA AMOSTRA DE PAciENTES COM A SÍNDROME DO OLHO DO GATO RAFAEL FABIANO MACHADO ROSA1, RÔMULO MOMBACH2, PAULO RicARDO GAZZOLA ZEN3, CARLA GRAZIADIO4, GIORGIO ADRIANO PASKULIN3* Trabalho realizado na Universidade Federal de Ciências da Saúde de Porto Alegre (UFCSPA) e Complexo Hospitalar Santa Casa de Porto Alegre (CHSCPA), Porto Alegre, RS RESUMO OBJETIVO. A síndrome do olho do gato é considerada uma doença cromossômica rara e fenotipicamente bastante variável. O objetivo deste trabalho foi descrever as características clínicas de uma amostra de pacientes com a síndrome avaliada em nosso serviço. MÉTODOS. Foram analisados, retrospectivamente, seis pacientes com diagnóstico de síndrome do olho do gato. Todos eles apresentavam cariótipo com a presença de um cromossomo marcador adicional, inv dup(22)(pter->q11.2::q11.2->pter). Um deles, ainda, possuía mosacismo com uma linhagem com constituição cromossômica normal. A partir dos prontuários médicos foram coletados dados clínicos e de evolução dos pacientes. Para comparação entre as frequências encontradas em nosso estudo e a literatura foi utilizado o teste exato de Fisher (P<0,05). RESULTADOS. As principais anormalidades encontradas foram os apêndices/fossetas pré-auriculares e a imperfuração anal (ambas observadas em 83% dos casos). O coloboma de íris, um achado importante da síndrome, foi verificado em dois casos (33%). Cardiopatia congênita, por sua vez, foi observada em quatro pacientes (67%), sendo o principal defeito a comunicação interatrial (75%). Achados incomuns incluíram a microssomia hemifacial associada à microtia, além da atresia de vias biliares. Quanto à evolução clínica, apenas um dos pacientes foi a óbito, sendo que este ocorreu secundário a um quadro de quilotórax e sepse. -

Easychip 8X15k
ndrom Sy es tic & e G n e e n G e f T o Alesi et al., J Genet Syndr Gene Ther 2016, 7:1 Journal of Genetic Syndromes h l e a r n a DOI: 10.4172/2157-7412.1000277 r p u y o J & Gene Therapy ISSN: 2157-7412 Research Article Open Access Easychip 8x15k: A New Tool for Detecting Chromosome Anomalies in Low Risk Pregnancies, Supporting and Integrating Standard Karyotype Viola Alesi1*, Laura Bernardini2 , Didier Goidin3 , Michela Canestrelli4 , Maria Lisa Dentici1 , Giuseppe Barrano4 , Maria Grazia Giuffrida2 , Anna Maria Nardone5 , Diana Postorivo5 , Luigi Laino6 , Rita Genesio7 , Bruno Dallapiccola1 and Antonio Novelli1 1Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy 2Mendel Laboratory, IRCSS Casa Sollievo della Sofferenza Hospital, Rome, Italy 3Genomics Group, Agilent Technologies, Les Ulis, France 4San Pietro Fatebenefratelli Hospital, Rome, Italy 5Fondazione Policlinico Tor Vergata, Rome, Italy 6Division of Medical Genetics, Department of Molecular Medicine, Sapienza University, San Camillo-Forlanini Hospital, Rome, Italy 7Department of Molecular Medicine and Medical Biotechnology, University of Naples “Federico II”, Naples, Italy Abstract Over last decade chromosome microarray analysis has become a routine test, but its use as first tier in prenatal diagnosis still raises disputes specially when applied to low risk pregnancies. In order to limit the identification of incidental findings (IF) and variants of unknown significance (VOUS) we designed EasyChip, a low-resolution oligonucleotide array CGH platform with a functional resolution of 3 Mb in genomic backbone, 300 Kb in sub-telomeric regions, and 150 Kb in 43 regions associated with syndromic disorders, selected considering morbidity, penetrance, and etiological mechanisms. -

Cat Eye Syndrome: Case Report
Tepecik Eğit. ve Araşt. Hast. Dergisi 2018;28(1):72-74 Olgu Sunumu doi:10.5222/terh.2018.072 Cat Eye syndrome: Case report Cat Eye sendromu: Olgu sunumu Özlem ÜZÜM1, Tuba TınasTEPE1, Kayı ElİaçıK1, Yaşar Bekir KutBaY2, Özgür KırBıYıK2, Berrak Sarıoğlu1 1Çocuk Sağlığı ve Hastalıkları, Tepecik Eğitim ve Araştırma Hastanesi, İzmir, Türkiye 2Genetik Tanı Merkezi, Tepecik Eğitim ve Araştırma Hastanesi, İzmir, Türkiye ABSTRACT Cat Eye Syndrome (CES) or Schmid-Fracccaro syndrome is a genetic disorder cha- racterized by mutations in the long arm of chromosome 22, first described by Schachenmann et al. in 1965. Its classic triad consists of iris coloboma, anal malfor- mation and ear anomalies. In this case, a 3-year-old male patient with a 30-second tonic clonic seizure was presented. On physical examination, he had downslanting palpebral fissures, micrognathia and microphthalmia. His family told that he was monitored with mild retardation in cognitive development by the department of pedi- atric psychiatry. Karyotypic analysis was performed for cat eye syndrome, because of the presence of neuropsychiatric findings and eye / facial anomalies. DNA microarray analysis revealed a gain involving 3 OMIM genes in the 22q11.1 region of about 548 Kb was detected. In this case, there were no major anomalies, but followed by genetic consequence analysis, and diagnosed with Cat-Eye Syndrome. Therefore, genetic analysis should be requested in case of clinical suspicion in atypical cases without classic triad. Keywords: Neuropsychiatric findings, atypical facial features, seizure, Cat Eye Syndrome ÖZ Cat Eye Sendromu (CES) veya Schmid-Fracccaro sendromu ilk kez 1965 yılında Schachenmann ve ark.