Insufficient FDA Resources: Levelling the Playing Field and Reducing Fraud by Altering Incentives
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Consumer Behavior & Marketing Strategy
e 99e f o noitidE htniN e hT T h e N i n t h E d i tion of a kool l o o k a ram m a r ahe bbe h a l anAAn a l f o o f aheb remusn oc ;noco i t ic on ggn i t i o n ; c o n s u m e r b eha r utcurts s t r u c t u r l ana re musno cco n s u m e r a n a l Ninth Edition CONSUMER BEHAVIOR & MARKETING STRATEGY J. Paul Peter Jerry C. Olson Consumer Behavior & Marketing Strategy Ninth Edition J. Paul Peter University of Wisconsin, Madison Jerry C. Olson Pennsylvania State University Olson Zaltman Associates CONSUMER BEHAVIOR & MARKETING STRATEGY Published by McGraw-Hill/Irwin, a business unit of The McGraw-Hill Companies, Inc., 1221 Avenue of the Americas, New York, NY, 10020. Copyright © 2010, 2008, 2005, 2002, 1999, 1996, 1993, 1990, 1987 by The McGraw-Hill Companies, Inc. All rights reserved. No part of this publication may be reproduced or distributed in any form or by any means, or stored in a database or retrieval system, without the prior written consent of The McGraw-Hill Companies, Inc., including, but not limited to, in any network or other electronic storage or transmission, or broadcast for distance learning. Some ancillaries, including electronic and print components, may not be available to customers outside the United States. This book is printed on acid-free paper. 1 2 3 4 5 6 7 8 9 0 CCW/CCW 0 9 ISBN 978-0-07-340476-9 MHID 0-07-340476-4 Vice president and editor-in-chief: Brent Gordon Publisher: Paul Ducham Director of development: Ann Torbert Managing development editor: Laura Hurst Spell Editorial assistant: Jane Beck Vice president and director of marketing: Robin J. -

Weekly World Ambulance Chaser, Centerfold
College of William & Mary Law School William & Mary Law School Scholarship Repository Student Newspaper (Amicus, Advocate...) Archives and Law School History 1993 Amicus Curiae (Vol. 3, Issue 11) Repository Citation "Amicus Curiae (Vol. 3, Issue 11)" (1993). Student Newspaper (Amicus, Advocate...). 325. https://scholarship.law.wm.edu/newspapers/325 Copyright c 1993 by the authors. This article is brought to you by the William & Mary Law School Scholarship Repository. https://scholarship.law.wm.edu/newspapers Weekly World Ambulance Chaser, centerfold. AMERICA'S FIRST LAW SCHOOL VOLUME Ill, ISSUE ELEVEN THURSDAY, APRIL 1,1993 TWENTY EIGHT-PAGES FDA Chief to Speak at M -W Graduation By PETER KUBIN other Bush appointees. " Who?" Many students at Kesslerholds degrees in both Marshall-Wythe echoed third law and medicine, and has been year Cathy Clemens' reaction to the FDA head since late 1990. the annOWlcement that David Upon his appointment, Kessler Kessler would be this year's law wasted no time launching a school commencement speaker. number of highly visible Kessler, who was appointed by enforcement actions, starting George Bush to head. the Food with the seizure of thou sands of and Drug Administration, has cases of "Citrus Hill " proven to be an aggressive and reconstituted orange juice that sometimes controversial carried the misleading label administrator of the agency. "fresh." The FDA has made Apparently garnering more news during his tenure a number respect in Washington than at of times, most notably for their M-W, Kesslerrecentlyaccepted overhaul of the federally an offer to retain his position, although he offered his See DRUGS, page 28 Carter O. -
UFDC Image Array 2
Project1:Layout 1 6/10/2014 1:13 PM Page 1 MLB: Tampa Bay takes on division rival Toronto /A10 SATURDAY TODAY CITRUSCOUNTY & next morning HIGH 92 Scattered LOW showers and storms. 72 PAGE A4 www.chronicleonline.com AUGUST 15, 2020 Florida’s Best Community Newspaper Serving Florida’s Best Community 50¢ VOL. 125 ISSUE 312 NEWS BRIEFS Legal battle over schools advances case doesn’t mean that the challenges to Citrus COVID-19 Judge refuses to toss out HIGH SCHOOL SPORTS ARE ON Corcoran’s July 6 emergency order will be daily update n The FHSAA on Friday voted that high successful. school athletic practices may begin During an hour-long video hearing, the Forty-four new positive lawsuits concerning Aug . 24, with Sept . 4 the first day games judge also encouraged attorneys represent- cases were reported in Cit- can be played . See Page A9 for story . ing the unions and the defendants — Gov. Ron rus County since the latest DeSantis, Corcoran and other state education FDOH update. One new reopening classrooms refused to toss out lawsuits challenging Edu- officials — to settle the dispute by hospitalization was re- Wednesday. DARA KAM cation Commissioner Richard Corcoran’s ported; no new deaths News Service of Florida mandate that school districts reopen class- “This is a case that cries out for the parties were reported. rooms this month. to get together at this mediation and come up To date in the county, TALLAHASSEE — Siding with teachers But Judge Charles Dodson said his deci- with an agreement. It’s a very complicated 1,703 people have tested unions, a Leon County circuit judge on Friday sion to deny the state’s motion to dismiss the See BATTLE/Page A5 positive (including five non-residents), 156 have been hospitalized and 38 have died. -

REACH out Address Touch Nation's Hands Udgrads Author of 'Roots' at Ceremony Sunday
LIBRA.R y JUN Newark students 'Just Say No' to drugs/6a Carolina wins lacrosse title/1 b Colonial. residents routed/3a Vol. 75, o. 51 May28,1986 Newark, Del. Newarkers Haley to REACH OUT address Touch nation's hands UDgrads Author of 'Roots' at ceremony Sunday The University of Delaware will hold its 137th Commencement ceremonies at 10 a.m., Sunday, June 1, in Delaware Stadium, rain or shine. No invitation is necessary, and anyone who wishes to attend may do so. Delaware Stadium is located on Del. 896 in Newark. Commencement speaker will be Alex Haley, acclaim ed author of "Roots." Before the Commence ment ceremony, graduating students, their parents and friends who wish to n1eet Univers.ty President E.A. Trabimt may do so from 9- 9:30,Ji .m . at the entrance to the west stands. Those planning to attend Commencement are advised to arrive by 8:30a.m., because of possible traffic congestion. Seating at Commence ment will be in the east and west stands. Limited on- field seating will be reserv \ ed for handicapped persons, and the first several rows of by Bruce Johnson the seats in the stands will be reserved for elderly per sons who are unable to keptics said it couldn't be done. negotiate the stadium steps. But when it was done, nearly Commencement activities five million smiling Americans will begin on Saturday, May S stretched a broken line from 31, when each of the Univer coast to coast and proved otherwise. sity's 10 colleges will hold Hands Across America was a big suc Convocation exercises on cess. -
Jobless System Swamped
Project1:Layout 1 6/10/2014INSIDE 1:13 PM Page 1 Big burgers, hot dog! lair for food / B1 Enjoy Memorial Day with the tradition of f meaty burgers and wonderful wieners on the grill. THURSDAY TODAY CITRUSCOUNTY & next morning HIGH 90 Sunshine, LOW becoming more humid. 66 PAGE A4 www.chronicleonline.com MAY 21, 2020 Florida’s Best Community Newspaper Serving Florida’s Best Community 50¢ VOL. 125 ISSUE 226 INSIDE LOCAL: Spotlight on Jobless system swamped business The Chronicle is DeSantis: Historic surge of unemployment claims overwhelmed computers coordinating spotlights on local businesses that JOHN HAUGHEY unemployment system “designed to fail.” simultaneously and could n For the latest on Citrus are adapting to the The Center Square after being confronted the “We had an unprece- boost-up to 5,000, but County unemployment coronavirus . To submit past week by reporters dented surge of unemploy- when the response to the Florida Gov. Ron DeSan- figures, see Saturday’s who said they’ve been del- ment claims. That was a COVID-19 emergency your own business edition of the Citrus spotlight, visit https:// tis said the state’s revamped uged by people demanding tough challenge under any forced businesses to shut unemployment system has County Chronicle . answers to why so many circumstance but even a down, it was over- bit .ly/2LsNIhU . For sent checks to 97.6% of n had not received unem- greater challenge that the whelmed by hundreds of today’s spotlight, see Experts point to need nearly 900,000 “processed” for reform ./Page A9 ployment payments. -

What Can the Price Gap Between Branded and Private Label
1%(5:25.,1*3$3(56(5,(6 :+$7&$17+(35,&(*$3%(7:((1%5$1'('$1'35,9$7(/$%(/352'8&76 7(//86$%2870$5.836" 5REHUW%DUVN\ 0DUN%HUJHQ 6KDQWDQX'XWWD 'DQLHO/HY\ :RUNLQJ3DSHU KWWSZZZQEHURUJSDSHUVZ 1$7,21$/%85($82)(&2120,&5(6($5&+ 0DVVD4KXVHWWV$YHQXH &DPEULGJH70$ $XJXVW :HDUHJUDWHIXOWRWKHSDSHU¶VGLVXVVDQW-XOLR5RWHPEHUJIRUKLVLQVLJKWIXORPPHQWVZKLKSOD\HGD IXQGDPHQWDOUROHLQWKHUHVWUXWXULQJDQGVWUHQJWKHQLQJRIWKHDUJXPHQW6XVDQWR%DVXDQG0DWWKHZ6KDSLUR SURYLGHGQXPHURXVYDOXDEOHVXJJHVWLRQVDQGHVVHQWLDODGYLH:HZRXOGOLNHWRWKDQNDOVR1LQJ/LXIRUKHU RXWVWDQGLQJUHVHDUKDVVLVWDQHXQGHUDYHU\WLJKWVKHGXOH2ZHQ%HHOGHUV(UQLH%HUQGW+DVKHP'H]KEDNKVK -LP+HVV.DLXZH.XKQ-LP/HYLQVRKQ3HWHU5RVVLDQG6WHYH6DODQWIRUPDQ\KHOSIXOGLVXVVLRQV5REHUW )HHQVWUDIRURPPHQWV6WHYH+RKIRUSURYLGLQJXVZLWKKLVVXUYH\GDWDRQQDWLRQDOEUDQGSULYDWHODEHOTXDOLW\ GLIIHUHQHV DQG -RQJ .LP IRU KLV KHOS ZLWK RPSXWDWLRQV :H WKDQN WKH SDUWLLSDQWV RI WKH 1%(5&5,: RQIHUHQHWKH0LGZHVW0DUNHWLQJ&DPSDQGVHPLQDUSDUWLLSDQWVDWWKH8QLYHUVLW\RI0LKLJDQDQGWKH 8QLYHUVLW\RI0LQQHVRWD)LQDOO\ZHWKDQNERWKWKH8QLYHUVLW\RI&KLDJRDQG'RPLQLN¶VIRUSURYLGLQJXVZLWK WKHGDWD7KHYLHZVH[SUHVVHGKHUHLQDUHWKRVHRIWKHDXWKRUVDQGQRWQHHVVDULO\WKRVHRIWKH1DWLRQDO%XUHDXRI (RQRPL5HVHDUK E\5REHUW%DUVN\0DUN%HUJHQ6KDQWDQX'XWWDDQG'DQLHO/HY\$OOULJKWVUHVHUYHG6KRUWVHWLRQV RIWH[WQRWWRH[HHGWZRSDUDJUDSKVPD\EHTXRWHGZLWKRXWH[SOLLWSHUPLVVLRQSURYLGHGWKDWIXOOUHGLW LQOXGLQJQRWLHLVJLYHQWRWKHVRXUH :KDW&DQWKH3UL4H*DSEHWZHHQ%UDQGHGDQG3ULYDWH/DEHO3URGX4WV 7HOO8VDERXW0DUNXSV" 5REHUW%DUVN\70DUN%HUJHQ76KDQWDQX'XWWDDQG'DQLHO/HY\ 1%(5:RUNLQJ3DSHU1R $XJXVW $%675$&7 ,QWKLVSDSHUZHLQYHVWLJDWHWKHVL]HRIPDUNXSVIRUQDWLRQDOO\EUDQGHGSURGX4WVVROGLQWKH86 -

The BG News September 1, 1989
Bowling Green State University ScholarWorks@BGSU BG News (Student Newspaper) University Publications 9-1-1989 The BG News September 1, 1989 Bowling Green State University Follow this and additional works at: https://scholarworks.bgsu.edu/bg-news Recommended Citation Bowling Green State University, "The BG News September 1, 1989" (1989). BG News (Student Newspaper). 4963. https://scholarworks.bgsu.edu/bg-news/4963 This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 4.0 License. This Article is brought to you for free and open access by the University Publications at ScholarWorks@BGSU. It has been accepted for inclusion in BG News (Student Newspaper) by an authorized administrator of ScholarWorks@BGSU. Friday Weather Vol.72 Issue 8 High 80° September 1, 1989 Low 65° Bowling Green, Ohio The BG News Regents BRIEFLY cut funds by John Chalfant State Associated Press writer Church code: At the Sacred COLUMBUS - An Ohio Board Heart Church in Dayton, if a member of Regents committee on Thurs- is seen wearing "immodest" clothing, day began reviewing tentative they will be denied communion. staff plans for cuts in a $1 billion statewide list of construction Examples include shorts, mini-skirts requests submitted by public un- and halter tops. iversities and colleges. Consideration by the panel OSU informs: If you have marked the first step in a pro- questions, The Ohio State University cess that will end in December has the answers. A new information when the regents submit a service "hotline" can refer you to scaled-down package of new experts at the university who can building and renovation projects answer the questions it cannot. -

Flat-Coated Retriever Society of America *Please Refer to Flat-Coated Retriever Society — “Camera Ready” Ad Requirements Inside
Specialty / Summer 2019 DEADLINES: Fall 2019 Issue: ........................................................... Thursday, October 10, 2019* Winter 2020 Issue: ............................................................Friday, January 10, 2020* Spring 2020 Issue: ..................................................................Friday, April 10, 2020* 3rd of month to qualify for ad discount ADVERTISING RATES: Full page Camera Ready/PDF* $40.00 (early discounted price - $34.00) Half page Camera Ready/PDF* $20.00 (early discounted price - $17.00) Full page, non Camera Ready* no photo $40.00 (no discount) Full page non Camera Ready* with photo $60.00 (no discount) Half page, non Camera Ready* no photo $20.00 (no discount) Half page non Camera Ready* with photo $30.00 (no discount) Additional photos (each) $10.00 (no discount) Litter listing, no ad $15.00 (early discounted price - $12.75) Litter listing with ad Included in ad price Commercial advertising -- members and non-members 3 times basic rates Flat-Coated Retriever Society of America *Please refer to Flat-Coated Retriever Society — “Camera Ready” Ad Requirements inside All photos returned promptly (please put address label on back of photos). The FCRSA requires that litter listings or advertisements for a single or multiple-sired breeding must include copies of OFA or equivalent hip and patella ratings and OFA, CERF, or ACVO eye certifications for the dam and sire or all sires. Eye exams must be within 12 months of breeding to be valid, as per FCRSA Board decision. Litter listings without determined (named) sire(s) will not be accepted. Include payment with ads. Checks payable to FCRSA, Inc. and send to Christine Teneralli, address inside front cover. Litter advertising, dogs for sale and stud dog ads accepted ONLY from FCRSA Members in good standing at the time of publication. -
Cadenza Document
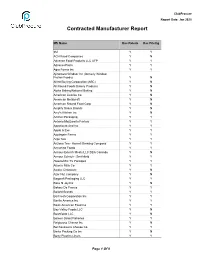
ClubProcure Report Date: Jan 2020 Contracted Manufacturer Report Mfr Name Has Rebate Has Pricing 3M Y Y ACH Food Companies Y N Advance Food Products LLC AFP Y Y AdvancePierre Y Y Agro Farma Inc Y Y Ajinomoto Windsor Inc (formerly Windsor Frozen Foods) Y N Allied Buying Corporation (ABC) Y N All Round Foods Bakery Products Y N Alpha Baking/National Baking Y N American Licorice Co Y N American Metalcraft Y N American Roland Food Corp Y N Amplify Snack Brands Y N Amy's Kitchen Inc Y N Anchor Packaging Y Y Antonio Mozzarella Factory Y Y Appetizers And Inc Y Y Apple & Eve Y Y Applegate Farms Y Y Argo Tea Y Y Arizona Tea - Hornell Brewing Company Y Y Armanino Foods Y Y Armour-Eckrich Meats LLC DBA Carando Y N Armour Eckrich - Smithfield Y Y Ateeco/Mrs T's Pierogies Y Y Atlantic Mills Co Y Y Awake Chocolate Y N Azar Nut Company Y N Bagcraft Packaging LLC Y Y Bake N Joy Inc Y N Bakery De France Y Y Ballard Brands Y Y BarFresh Corporation Inc Y Y Barilla America Inc Y Y Basic American Food Co Y Y Bay Valley Foods LLC Y N Beanfields LLC Y N Beaver Street Fisheries Y Y Belgioioso Cheese Inc Y N Bel Kaukauna Cheese Co Y Y Berks Packing Co Inc Y N Berry Plastics Liners Y Y Page 1 Of 9 ClubProcure Report Date: Jan 2020 Contracted Manufacturer Report Mfr Name Has Rebate Has Pricing Beverage Air Y N Beyond Meat Y Y B&G Foods Inc Y Y Biena Snacks Y N BioSelect N Y Biscomerica Corp. -

Coppead Graduate School of Business the Federal University of Rio De Janeiro (Ufrj) Thiago Guedes De Melo How Growth May Affect
COPPEAD GRADUATE SCHOOL OF BUSINESS THE FEDERAL UNIVERSITY OF RIO DE JANEIRO (UFRJ) THIAGO GUEDES DE MELO HOW GROWTH MAY AFFECT THE SURVIVAL OF LONG-LIVED ORGANIZATIONS: THE COLGATE AND PROCTER & GAMBLE CASES RIO DE JANEIRO 2016 THIAGO GUEDES DE MELO HOW GROWTH MAY AFFECT THE SURVIVAL OF LONG-LIVED ORGANIZATIONS: THE COLGATE AND PROCTER & GAMBLE CASES Thesis submitted to the Instituto COPPEAD de Administração, Universidade Federal do Rio de Janeiro (UFRJ) as a prerequisite for the completion of the Full-time MBA program and the obtention of a M.Sc. title. Supervisor: Professor: Denise Lima Fleck, Ph.D. RIO DE JANEIRO 2016 2 3 THIAGO GUEDES DE MELO HOW GROWTH MAY AFFECT THE SURVIVAL OF LONG-LIVED ORGANIZATIONS: THE COLGATE AND PROCTER & GAMBLE CASES Thesis submitted to the Instituto COPPEAD de Administração, Universidade Federal do Rio de Janeiro (UFRJ) as a prerequisite for the completion of the Full-time MBA program and the obtention of a M.Sc. title. Approved by: ____________________________________________________ Prof. Denise Lima Fleck, Ph.D (COPPEAD, UFRJ) - Supervisor ____________________________________________________ Prof. Paula Chimenti, Ph.D (COPPEAD, UFRJ) ____________________________________________________ Prof. José Vitor Bomtempo Martins, Ph.D (PUC-Rio) RIO DE JANEIRO 2016 4 ACKNOWLEDGEMNTS To my grandparents, Adélia and Fernando Guedes, Denise and Maurilio de Melo, who have set the roots of a united and supportive Family. To my parents, Laurinda and Murilo Melo, who have shown me how to live, love, laugh and learn. To my brother and best friend, Rafael Melo, who has shared with me so many adventures. To my better-half, Natalia Muniz, who has brightened my world. -

Australian Customs Notice No. 2005/41
AUSTRALIAN CUSTOMS NOTICE NO. 2005/41 Notices of Objection to Importation Trade Marks Act 1995 The Trade Marks Act 1995 allows the registered owner, or in certain circumstances, the authorised user of a trademark to object to the importation of goods which infringe their trademark. The registered owner, or authorised user does this by lodging a Notice of Objection with the Australian Customs Service (Customs). Unless revoked, a Notice of Objection remains in force for a period of two years from the date of commencement. The attached Schedule sets out the registered owners and authorised users who have lodged Notices of Objection under the Trade Marks Act 1995 since July 2005 (ACN 2005/33). The Notices notify the objections of these registered owners and authorised users to the importation of goods which infringe their trade mark or trade marks. Descriptions of the relevant trade marks are also set out in the Schedule. A detailed list of all Notices of Objection is available at http://cww.customs.gov.au/internet/site/page4683.htm. For each particular trade mark described, the Schedule makes reference to a particular “class” of goods. This refers to the classes of goods prescribed in Schedule 1 to the Trade Marks Regulations 1995 and along with a description of the goods, is used to describe the type of goods for which the particular trade mark is registered. Prospective importers of such goods should seek further advice from Customs regarding the ambit of any Notice of Objection set out in the Schedule. Where goods of the kind referred to manufactured outside and imported into Australia: − have applied to them, or in relation to them, a sign that is substantially identical with, or deceptively similar to, a trade mark listed in the Schedule; and − fall within a class of goods for which the relevant trade mark is registered, they are liable to be seized by Customs unless it can be established that: − the goods are being imported otherwise than for the purposes of trade (sale, lease, hire, etc.); or − the goods do not infringe the relevant trade mark. -

Riverside County Public Schools Directory
RIVERSIDE COUNTY PUBLIC SCHOOLS DIRECTORY 2010-2011 Published by the: Riverside County Office of Education 3939 Thirteenth Street, P.O. Box 868 Riverside, California 92502-0868 (951) 826-6530 www.rcoe.us (Note: Most of Riverside County is in the 951 area code unless otherwise noted.) 2 OUR PLEDGE All students in Riverside County will graduate from high school well prepared for college and the workforce. Mission The mission of RCOE is to ensure the success of all students through extraordinary service, support, and partnerships. Vision RCOE will be a collaborative organization characterized by the highest quality employees providing leadership, programs and services to school districts, schools, and students countywide. Core Values Engage in Open and Honest Communication Build Relationships and Promote Trust Focus on Students and Children 1 Historical Note: The first Riverside County Schools Directory was published in 1901 by the Riverside County Superintendent of Schools. It consisted of a two‐page listing of 69 teachers in 69 schools. In the 109th edition of the Riverside County Public Schools Directory, there are listings for over 486 schools serving 431,373 K‐12 students. There are more than 18,742 teachers and 17,476 non‐teaching school employees serving students, families, and the communities of Riverside County. Corrections: Should you notice an error in the directory or have any changes to the current listings, please notify the Office of the Riverside County Superintendent of Schools at (951) 826‐6674 or email [email protected]. Riverside County Office of Education ADMINISTRATIVE OFFICES Riverside - Main Office 3939 Thirteenth Street/P.O.