An Overview of Self-Consistent Field Calculations Within Finite Basis Sets †
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Atomic Orbitals
Visualization of Atomic Orbitals Source: Robert R. Gotwals, Jr., Department of Chemistry, North Carolina School of Science and Mathematics, Durham, NC, 27705. Email: [email protected]. Copyright ©2007, all rights reserved, permission to use for non-commercial educational purposes is granted. Introductory Readings Where are the electrons in an atom? There are a number of Models - a description of models that look to describe the location and behavior of some physical object or electrons in atoms. It is important to know this information, as behavior in nature. Models it helps chemists to make statements and predictions about are often mathematical, describing the behavior of how atoms will react with other atoms to form molecules. The some event. model one chooses will oftentimes influence the types of statements one can make about the behavior – that is, the reactivity – of an atom or group of atoms. Bohr model - states that no One model, the Bohr model, describes electrons as small two electrons in an atom can billiard balls, rotating around the positively charged nucleus, have the same set of four much in the way that planets quantum numbers. orbit around the Sun. The placement and motion of electrons are found in fixed and quantifiable levels called orbits, Orbits- where electrons are or, in older terminology, shells. believed to be located in the There are specific numbers of Bohr model. Orbits have a electrons that can be found in fixed amount of energy, and are identified with the each orbit – 2 for the first orbit, principle quantum number 8 for the second, and so forth. -
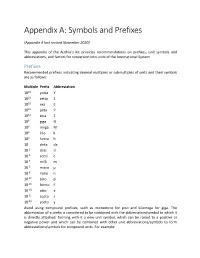
Appendix A: Symbols and Prefixes
Appendix A: Symbols and Prefixes (Appendix A last revised November 2020) This appendix of the Author's Kit provides recommendations on prefixes, unit symbols and abbreviations, and factors for conversion into units of the International System. Prefixes Recommended prefixes indicating decimal multiples or submultiples of units and their symbols are as follows: Multiple Prefix Abbreviation 1024 yotta Y 1021 zetta Z 1018 exa E 1015 peta P 1012 tera T 109 giga G 106 mega M 103 kilo k 102 hecto h 10 deka da 10-1 deci d 10-2 centi c 10-3 milli m 10-6 micro μ 10-9 nano n 10-12 pico p 10-15 femto f 10-18 atto a 10-21 zepto z 10-24 yocto y Avoid using compound prefixes, such as micromicro for pico and kilomega for giga. The abbreviation of a prefix is considered to be combined with the abbreviation/symbol to which it is directly attached, forming with it a new unit symbol, which can be raised to a positive or negative power and which can be combined with other unit abbreviations/symbols to form abbreviations/symbols for compound units. For example: 1 cm3 = (10-2 m)3 = 10-6 m3 1 μs-1 = (10-6 s)-1 = 106 s-1 1 mm2/s = (10-3 m)2/s = 10-6 m2/s Abbreviations and Symbols Whenever possible, avoid using abbreviations and symbols in paragraph text; however, when it is deemed necessary to use such, define all but the most common at first use. The following is a recommended list of abbreviations/symbols for some important units. -

Chemistry 2000 Slide Set 1: Introduction to the Molecular Orbital Theory
Chemistry 2000 Slide Set 1: Introduction to the molecular orbital theory Marc R. Roussel January 2, 2020 Marc R. Roussel Introduction to molecular orbitals January 2, 2020 1 / 24 Review: quantum mechanics of atoms Review: quantum mechanics of atoms Hydrogenic atoms The hydrogenic atom (one nucleus, one electron) is exactly solvable. The solutions of this problem are called atomic orbitals. The square of the orbital wavefunction gives a probability density for the electron, i.e. the probability per unit volume of finding the electron near a particular point in space. Marc R. Roussel Introduction to molecular orbitals January 2, 2020 2 / 24 Review: quantum mechanics of atoms Review: quantum mechanics of atoms Hydrogenic atoms (continued) Orbital shapes: 1s 2p 3dx2−y 2 3dz2 Marc R. Roussel Introduction to molecular orbitals January 2, 2020 3 / 24 Review: quantum mechanics of atoms Review: quantum mechanics of atoms Multielectron atoms Consider He, the simplest multielectron atom: Electron-electron repulsion makes it impossible to solve for the electronic wavefunctions exactly. A fourth quantum number, ms , which is associated with a new type of angular momentum called spin, enters into the theory. 1 1 For electrons, ms = 2 or − 2 . Pauli exclusion principle: No two electrons can have identical sets of quantum numbers. Consequence: Only two electrons can occupy an orbital. Marc R. Roussel Introduction to molecular orbitals January 2, 2020 4 / 24 The hydrogen molecular ion The quantum mechanics of molecules + H2 is the simplest possible molecule: two nuclei one electron Three-body problem: no exact solutions However, the nuclei are more than 1800 time heavier than the electron, so the electron moves much faster than the nuclei. -

Theoretical Methods That Help Understanding the Structure and Reactivity of Gas Phase Ions
International Journal of Mass Spectrometry 240 (2005) 37–99 Review Theoretical methods that help understanding the structure and reactivity of gas phase ions J.M. Merceroa, J.M. Matxaina, X. Lopeza, D.M. Yorkb, A. Largoc, L.A. Erikssond,e, J.M. Ugaldea,∗ a Kimika Fakultatea, Euskal Herriko Unibertsitatea, P.K. 1072, 20080 Donostia, Euskadi, Spain b Department of Chemistry, University of Minnesota, 207 Pleasant St. SE, Minneapolis, MN 55455-0431, USA c Departamento de Qu´ımica-F´ısica, Universidad de Valladolid, Prado de la Magdalena, 47005 Valladolid, Spain d Department of Cell and Molecular Biology, Box 596, Uppsala University, 751 24 Uppsala, Sweden e Department of Natural Sciences, Orebro¨ University, 701 82 Orebro,¨ Sweden Received 27 May 2004; accepted 14 September 2004 Available online 25 November 2004 Abstract The methods of the quantum electronic structure theory are reviewed and their implementation for the gas phase chemistry emphasized. Ab initio molecular orbital theory, density functional theory, quantum Monte Carlo theory and the methods to calculate the rate of complex chemical reactions in the gas phase are considered. Relativistic effects, other than the spin–orbit coupling effects, have not been considered. Rather than write down the main equations without further comments on how they were obtained, we provide the reader with essentials of the background on which the theory has been developed and the equations derived. We committed ourselves to place equations in their own proper perspective, so that the reader can appreciate more profoundly the subtleties of the theory underlying the equations themselves. Finally, a number of examples that illustrate the application of the theory are presented and discussed. -
![Arxiv:1809.04476V2 [Physics.Chem-Ph] 18 Oct 2018 (Sub)States](https://docslib.b-cdn.net/cover/8692/arxiv-1809-04476v2-physics-chem-ph-18-oct-2018-sub-states-198692.webp)
Arxiv:1809.04476V2 [Physics.Chem-Ph] 18 Oct 2018 (Sub)States
Quantum System Partitioning at the Single-Particle Level Quantum System Partitioning at the Single-Particle Level Adrian H. M¨uhlbach and Markus Reihera) ETH Z¨urich, Laboratorium f¨ur Physikalische Chemie, Vladimir-Prelog-Weg 2, CH-8093 Z¨urich,Switzerland (Dated: 17 October 2018) We discuss the partitioning of a quantum system by subsystem separation through unitary block- diagonalization (SSUB) applied to a Fock operator. For a one-particle Hilbert space, this separation can be formulated in a very general way. Therefore, it can be applied to very different partitionings ranging from those driven by features in the molecular structure (such as a solute surrounded by solvent molecules or an active site in an enzyme) to those that aim at an orbital separation (such as core-valence separation). Our framework embraces recent developments of Manby and Miller as well as older ones of Huzinaga and Cantu. Projector-based embedding is simplified and accelerated by SSUB. Moreover, it directly relates to decoupling approaches for relativistic four-component many-electron theory. For a Fock operator based on the Dirac one-electron Hamiltonian, one would like to separate the so-called positronic (negative-energy) states from the electronic bound and continuum states. The exact two-component (X2C) approach developed for this purpose becomes a special case of the general SSUB framework and may therefore be viewed as a system- environment decoupling approach. Moreover, for SSUB there exists no restriction with respect to the number of subsystems that are generated | in the limit, decoupling of all single-particle states is recovered, which represents exact diagonalization of the problem. -

A New Scalable Parallel Algorithm for Fock Matrix Construction
A New Scalable Parallel Algorithm for Fock Matrix Construction Xing Liu Aftab Patel Edmond Chow School of Computational Science and Engineering College of Computing, Georgia Institute of Technology Atlanta, Georgia, 30332, USA [email protected], [email protected], [email protected] Abstract—Hartree-Fock (HF) or self-consistent field (SCF) ber of independent tasks and using a centralized dynamic calculations are widely used in quantum chemistry, and are scheduler for scheduling them on the available processes. the starting point for accurate electronic correlation methods. Communication costs are reduced by defining tasks that Existing algorithms and software, however, may fail to scale for large numbers of cores of a distributed machine, particularly in increase data reuse, which reduces communication volume, the simulation of moderately-sized molecules. In existing codes, and consequently communication cost. This approach suffers HF calculations are divided into tasks. Fine-grained tasks are from a few problems. First, because scheduling is completely better for load balance, but coarse-grained tasks require less dynamic, the mapping of tasks to processes is not known communication. In this paper, we present a new parallelization in advance, and data reuse suffers. Second, the centralized of HF calculations that addresses this trade-off: we use fine- grained tasks to balance the computation among large numbers dynamic scheduler itself could become a bottleneck when of cores, but we also use a scheme to assign tasks to processes to scaling up to a large system [14]. Third, coarse tasks reduce communication. We specifically focus on the distributed which reduce communication volume may lead to poor load construction of the Fock matrix arising in the HF algorithm, balance when the ratio of tasks to processes is close to one. -

Domain Decomposition and Electronic Structure Computations: a Promising Approach
Domain Decomposition and Electronic Structure Computations: A Promising Approach G. Bencteux1,4, M. Barrault1, E. Canc`es2,4, W. W. Hager3, and C. Le Bris2,4 1 EDF R&D, 1 avenue du G´en´eral de Gaulle, 92141 Clamart Cedex, France {guy.bencteux,maxime.barrault}@edf.fr 2 CERMICS, Ecole´ Nationale des Ponts et Chauss´ees, 6 & 8, avenue Blaise Pascal, Cit´eDescartes, 77455 Marne-La-Vall´ee Cedex 2, France, {cances,lebris}@cermics.enpc.fr 3 Department of Mathematics, University of Florida, Gainesville, FL 32611-8105, USA, [email protected] 4 INRIA Rocquencourt, MICMAC project, Domaine de Voluceau, B.P. 105, 78153 Le Chesnay Cedex, FRANCE Summary. We describe a domain decomposition approach applied to the spe- cific context of electronic structure calculations. The approach has been introduced in [BCH06]. We survey here the computational context, and explain the peculiari- ties of the approach as compared to problems of seemingly the same type in other engineering sciences. Improvements of the original approach presented in [BCH06], including algorithmic refinements and effective parallel implementation, are included here. Test cases supporting the interest of the method are also reported. It is our pleasure and an honor to dedicate this contribution to Olivier Pironneau, on the occasion of his sixtieth birthday. With admiration, respect and friendship. 1 Introduction and Motivation 1.1 General Context Numerical simulation is nowadays an ubiquituous tool in materials science, chemistry and biology. Design of new materials, irradiation induced damage, drug design, protein folding are instances of applications of numerical sim- ulation. For convenience we now briefly present the context of the specific computational problem under consideration in the present article. -

Introduction to Molecular Orbital Theory
Chapter 2: Molecular Structure and Bonding Bonding Theories 1. VSEPR Theory 2. Valence Bond theory (with hybridization) 3. Molecular Orbital Theory ( with molecualr orbitals) To date, we have looked at three different theories of molecular boning. They are the VSEPR Theory (with Lewis Dot Structures), the Valence Bond theory (with hybridization) and Molecular Orbital Theory. A good theory should predict physical and chemical properties of the molecule such as shape, bond energy, bond length, and bond angles.Because arguments based on atomic orbitals focus on the bonds formed between valence electrons on an atom, they are often said to involve a valence-bond theory. The valence-bond model can't adequately explain the fact that some molecules contains two equivalent bonds with a bond order between that of a single bond and a double bond. The best it can do is suggest that these molecules are mixtures, or hybrids, of the two Lewis structures that can be written for these molecules. This problem, and many others, can be overcome by using a more sophisticated model of bonding based on molecular orbitals. Molecular orbital theory is more powerful than valence-bond theory because the orbitals reflect the geometry of the molecule to which they are applied. But this power carries a significant cost in terms of the ease with which the model can be visualized. One model does not describe all the properties of molecular bonds. Each model desribes a set of properties better than the others. The final test for any theory is experimental data. Introduction to Molecular Orbital Theory The Molecular Orbital Theory does a good job of predicting elctronic spectra and paramagnetism, when VSEPR and the V-B Theories don't. -

Matrix Algebra for Quantum Chemistry
Matrix Algebra for Quantum Chemistry EMANUEL H. RUBENSSON Doctoral Thesis in Theoretical Chemistry Stockholm, Sweden 2008 Matrix Algebra for Quantum Chemistry Doctoral Thesis c Emanuel Härold Rubensson, 2008 TRITA-BIO-Report 2008:23 ISBN 978-91-7415-160-2 ISSN 1654-2312 Printed by Universitetsservice US AB, Stockholm, Sweden 2008 Typeset in LATEX by the author. Abstract This thesis concerns methods of reduced complexity for electronic structure calculations. When quantum chemistry methods are applied to large systems, it is important to optimally use computer resources and only store data and perform operations that contribute to the overall accuracy. At the same time, precarious approximations could jeopardize the reliability of the whole calcu- lation. In this thesis, the selfconsistent eld method is seen as a sequence of rotations of the occupied subspace. Errors coming from computational ap- proximations are characterized as erroneous rotations of this subspace. This viewpoint is optimal in the sense that the occupied subspace uniquely denes the electron density. Errors should be measured by their impact on the over- all accuracy instead of by their constituent parts. With this point of view, a mathematical framework for control of errors in HartreeFock/KohnSham calculations is proposed. A unifying framework is of particular importance when computational approximations are introduced to eciently handle large systems. An important operation in HartreeFock/KohnSham calculations is the calculation of the density matrix for a given Fock/KohnSham matrix. In this thesis, density matrix purication is used to compute the density matrix with time and memory usage increasing only linearly with system size. The forward error of purication is analyzed and schemes to control the forward error are proposed. -

Guide for the Use of the International System of Units (SI)
Guide for the Use of the International System of Units (SI) m kg s cd SI mol K A NIST Special Publication 811 2008 Edition Ambler Thompson and Barry N. Taylor NIST Special Publication 811 2008 Edition Guide for the Use of the International System of Units (SI) Ambler Thompson Technology Services and Barry N. Taylor Physics Laboratory National Institute of Standards and Technology Gaithersburg, MD 20899 (Supersedes NIST Special Publication 811, 1995 Edition, April 1995) March 2008 U.S. Department of Commerce Carlos M. Gutierrez, Secretary National Institute of Standards and Technology James M. Turner, Acting Director National Institute of Standards and Technology Special Publication 811, 2008 Edition (Supersedes NIST Special Publication 811, April 1995 Edition) Natl. Inst. Stand. Technol. Spec. Publ. 811, 2008 Ed., 85 pages (March 2008; 2nd printing November 2008) CODEN: NSPUE3 Note on 2nd printing: This 2nd printing dated November 2008 of NIST SP811 corrects a number of minor typographical errors present in the 1st printing dated March 2008. Guide for the Use of the International System of Units (SI) Preface The International System of Units, universally abbreviated SI (from the French Le Système International d’Unités), is the modern metric system of measurement. Long the dominant measurement system used in science, the SI is becoming the dominant measurement system used in international commerce. The Omnibus Trade and Competitiveness Act of August 1988 [Public Law (PL) 100-418] changed the name of the National Bureau of Standards (NBS) to the National Institute of Standards and Technology (NIST) and gave to NIST the added task of helping U.S. -

Chemical Bonding & Chemical Structure
Chemistry 201 – 2009 Chapter 1, Page 1 Chapter 1 – Chemical Bonding & Chemical Structure ings from inside your textbook because I normally ex- Getting Started pect you to read the entire chapter. 4. Finally, there will often be a Supplement that con- If you’ve downloaded this guide, it means you’re getting tains comments on material that I have found espe- serious about studying. So do you already have an idea cially tricky. Material that I expect you to memorize about how you’re going to study? will also be placed here. Maybe you thought you would read all of chapter 1 and then try the homework? That sounds good. Or maybe you Checklist thought you’d read a little bit, then do some problems from the book, and just keep switching back and forth? That When you have finished studying Chapter 1, you should be sounds really good. Or … maybe you thought you would able to:1 go through the chapter and make a list of all of the impor- tant technical terms in bold? That might be good too. 1. State the number of valence electrons on the following atoms: H, Li, Na, K, Mg, B, Al, C, Si, N, P, O, S, F, So what point am I trying to make here? Simply this – you Cl, Br, I should do whatever you think will work. Try something. Do something. Anything you do will help. 2. Draw and interpret Lewis structures Are some things better to do than others? Of course! But a. Use bond lengths to predict bond orders, and vice figuring out which study methods work well and which versa ones don’t will take time. -

Electron Configurations, Orbital Notation and Quantum Numbers
5 Electron Configurations, Orbital Notation and Quantum Numbers Electron Configurations, Orbital Notation and Quantum Numbers Understanding Electron Arrangement and Oxidation States Chemical properties depend on the number and arrangement of electrons in an atom. Usually, only the valence or outermost electrons are involved in chemical reactions. The electron cloud is compartmentalized. We model this compartmentalization through the use of electron configurations and orbital notations. The compartmentalization is as follows, energy levels have sublevels which have orbitals within them. We can use an apartment building as an analogy. The atom is the building, the floors of the apartment building are the energy levels, the apartments on a given floor are the orbitals and electrons reside inside the orbitals. There are two governing rules to consider when assigning electron configurations and orbital notations. Along with these rules, you must remember electrons are lazy and they hate each other, they will fill the lowest energy states first AND electrons repel each other since like charges repel. Rule 1: The Pauli Exclusion Principle In 1925, Wolfgang Pauli stated: No two electrons in an atom can have the same set of four quantum numbers. This means no atomic orbital can contain more than TWO electrons and the electrons must be of opposite spin if they are to form a pair within an orbital. Rule 2: Hunds Rule The most stable arrangement of electrons is one with the maximum number of unpaired electrons. It minimizes electron-electron repulsions and stabilizes the atom. Here is an analogy. In large families with several children, it is a luxury for each child to have their own room.