Cardiovascular Mechanics2002.Kp
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cardiac Physiologyc As a Country
Type to enter text CardiacCardiovascular PhysiologyC as a Physiology Country Doc EpisodeEpisode 2:2: The EKG Electrocardiography I Patrick Eggena, M.D. Novateur Medmedia Type EPISODEto enter text 2 Type to enter text THE EKG by Patrick Eggena, M.D. i Copyright This Episode is derived from: Course in Cardiovascular Physiology by Patrick Eggena, M.D. © Copyright Novateur Medmedia, LLC. April 13, 2012 The United States Copyright Registration Number: PAu3-662-048 Ordering Information via iBooks: ISBN 978-0-9905771-1-9 Cardiac Physiology as a Country Doc, Episode 2: The EKG. Contact Information: Novateur Medmedia, LLC 39 Terry Hill Road, Carmel, NY 10512 email: [email protected] Credits: Oil Paintings by Bonnie Eggena, PsD. Music by Alan Goodman from his CD Under the Bed, Cancoll Music, copyright 2005 (with permission). Illustrations, movies, text, and lectures by Patrick Eggena, M.D. Note: Knowledge in the basic and clinical sciences is constantly changing. The reader is advised to carefully consult the instruc- tions and informational material included in the package inserts of each drug or therapeutic agent before administration. The Country Doctor Series illustrates Physiological Principles and is not intended as a guide to Medical Therapeutics. Care has been taken to present correct information in this book, however, the author and publisher are not responsible for errors or omissions or for any consequence from application of the information in this work and make no warranty, expressed or implied, with re- spect to the contents of this publication or that its operation will be uninterrupted and error free on any particular recording de- vice. -

Insights from Computational Modeling Into the Contribution of Mechano-Calcium Feedback on the Cardiac End-Systolic Force-Length Relationship
fphys-11-00587 May 28, 2020 Time: 19:46 # 1 ORIGINAL RESEARCH published: 29 May 2020 doi: 10.3389/fphys.2020.00587 Insights From Computational Modeling Into the Contribution of Mechano-Calcium Feedback on the Cardiac End-Systolic Force-Length Relationship Megan E. Guidry1, David P. Nickerson1, Edmund J. Crampin2,3, Martyn P. Nash1,4, Denis S. Loiselle1,5 and Kenneth Tran1* 1 Auckland Bioengineering Institute, The University of Auckland, Auckland, New Zealand, 2 Systems Biology Laboratory, School of Mathematics and Statistics, The University of Melbourne, Melbourne, VIC, Australia, 3 ARC Centre of Excellence in Convergent Bio-Nano Science and Technology, School of Chemical and Biomedical Engineering, The University of Melbourne, Melbourne, VIC, Australia, 4 Department of Engineering Science, The University of Auckland, Auckland, Edited by: New Zealand, 5 Department of Physiology, The University of Auckland, Auckland, New Zealand Leonid Katsnelson, Institute of Immunology and Physiology (RAS), Russia In experimental studies on cardiac tissue, the end-systolic force-length relation (ESFLR) Reviewed by: has been shown to depend on the mode of contraction: isometric or isotonic. The Andrey K. Tsaturyan, Lomonosov Moscow State University, isometric ESFLR is derived from isometric contractions spanning a range of muscle Russia lengths while the isotonic ESFLR is derived from shortening contractions across a Aurore Lyon, range of afterloads. The ESFLR of isotonic contractions consistently lies below its Maastricht University, Netherlands isometric counterpart. Despite the passing of over a hundred years since the first insight *Correspondence: Kenneth Tran by Otto Frank, the mechanism(s) underlying this protocol-dependent difference in the [email protected] ESFLR remain incompletely explained. -

Strategic Review of Cardiac Physiology Services in England: Final Report
STRATEGIC REVIEW OF CARDIAC PHYSIOLOGY SERVICES IN ENGLAND: FINAL REPORT Strategic Review of Cardiac Physiology Services in England Page 1 Final Report: 12/05/2015 CONTENTS Page Foreword 3 Summary of recommendations 4 Background 7 Purpose of the Review 8 Working arrangements and governance 8 Delivering a service model for the future 8 Delivering a cardiac physiology service to quality standards 16 Developing a cardiac physiology workforce for the future 18 Commissioning for quality 20 Appendix 1: 23 Cardiac physiology services in summary Appendix 2: 27 Working arrangements and governance Appendix 3: 28 Award in Practical Electrocardiography (APECG) Appendix 4: 29 Proposed possible key performance indicators Appendix 5: 30 Current status of standards development across a range of cardiac physiology investigations Appendix 6: 32 Current training, accreditation and standards Appendix 7: 33 National Review of Cardiac Physiology Services - Modelling the Future Workforce References 34 Glossary 36 Acknowledgements 38 Strategic Review of Cardiac Physiology Services in England Page 2 Final Report: 12/05/2015 FOREWORD In 2013, the Department of Health published the Cardiovascular Outcomes Strategy, a call to action to the health and care systems, which set out ten key actions to deliver for patients with cardiovascular disease improvements in their health outcomes. Cardiac physiology services are key to delivering the improved treatment and care, which is the ambition of the Outcomes Strategy. This Strategic Review of Cardiac Physiology Services was set up to make recommendations to NHS England, Health Education England and across the health and care system about the innovations in service delivery, models of care and workforce configuration which are needed to transform patients’ experience of cardiovascular disease care. -

Cardiac Physiology
Chapter 20 Cardiac Physiology LENA S. SUN • JOHANNA SCHWARZENBERGER • RADHIKA DINAVAHI K EY P OINTS • The cardiac cycle is the sequence of electrical and mechanical events during the course of a single heartbeat. • Cardiac output is determined by the heart rate, myocardial contractility, and preload and afterload. • The majority of cardiomyocytes consist of myofibrils, which are rodlike bundles that form the contractile elements within the cardiomyocyte. • The basic working unit of contraction is the sarcomere. • Gap junctions are responsible for the electrical coupling of small molecules between cells. • Action potentials have four phases in the heart. • The key player in cardiac excitation-contraction coupling is the ubiquitous second messenger calcium. • Calcium-induced sparks are spatially and temporally patterned activations of localized calcium release that are important for excitation-contraction coupling and regulation of automaticity and contractility. • β -Adrenoreceptors stimulate chronotropy, inotropy, lusitropy, and dromotropy. • Hormones with cardiac action can be synthesized and secreted by cardiomyocytes or produced by other tissues and delivered to the heart. • Cardiac reflexes are fast-acting reflex loops between the heart and central nervous system that contribute to the regulation of cardiac function and the maintenance of physiologic homeostasis. In 1628, English physician, William Harvey, first advanced oxygen (O2) and nutrients and to remove carbon dioxide the modern concept of circulation with the heart as the (CO2) and metabolites from various tissue beds. generator for the circulation. Modern cardiac physiology includes not only physiology of the heart as a pump but also concepts of cellular and molecular biology of the car- PHYSIOLOGY OF THE INTACT HEART diomyocyte and regulation of cardiac function by neural and humoral factors. -

Determination of Cardiac Output by Equating Venous Return Curves with Cardiac Response Curves
Determination of Cardiac Output By Equating Ve- nous Return Curves With Cardiac Response Curves1 ARTHUR C. GUYTQN From the Department of Physiology and Biophysics, School of Medicine, University of Mississippi, University, Mississippi HE CONCEPT that the heart responds with increasing cardiac output when there occurs increasing venous return was popularized by Starling and, in- deed, has come to be known as Starling’s law. There are many different forms in which Starling’s law can be expressed,including the relationship of cardiac output to right atria1 pressure, the relationship of cardiac output to the degree of distention of the right ventricle at the end of diastole, the relationship of cardiac work to right atria1 pressure or right ventricular distention, the relationship of left ventricular work to right atria1 pressure or right ventricular distention, etc. For the determina- tion of cardiac output, the form of Starling’s law which will be used in the present discussion is the relationship of cardiac output to mean right atria1 pressure, and this type of cruve will be called the “cardiac responsecurve” to right atria1 pressure. It is well known that many factors in the peripheral circulatory system combine together to determine the rate of venous return to the heart. These include the quantity of blood available, the degree of vascular resistance in various parts of the peripheral circulatory system, and the back pressure from the right atrium. It is with these factors that this paper is especially concerned, and it is hoped that this presentation will demonstrate how cardiac output is determined by equating the peripheral circulatory factors with the cardiac responsecurves. -
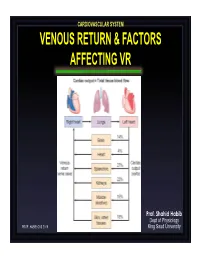
Venous Return & Factors Affecting Vr
CARDIOVASCULAR SYSTEM VENOUS RETURN & FACTORS AFFECTING VR Prof. Shahid Habib Dept of Physiology PROF. HABIB CVS 2019 King Saud University OBJECTIVES At the end of the lecture you should be able to ….. Discuss functions of the veins as blood reservoirs. Describe measurement of central venous pressure (CVP) and state its physiological and clinical significance. State determinants of venous return and explain how they influence venous return. Define mean systemic filling pressure, give its normal value and describe the factors which affect it. Explain the effect of gravity on venous pressure and explain pathophysiology of varicose veins. Describe vascular and cardiac function curves under physiological and pathophysiological conditions. DISTRIBUTION OF BLOOD Capacitance Vessels Veins are blood reservoirs When the body is at rest and many of the capillaries are closed, the capacity of the venous reservoir is increased as extra blood bypasses the capillaries and enters the veins. When this extra volume of blood stretches the veins, the blood moves forward through the veins more slowly because the total cross sectional area of the veins has increased as a result of the stretching. Therefore, blood spends more time in the veins. When the stored blood is needed, such as during exercise, extrinsic factors reduce the capacity of the venous reservoir and drive the extra blood from the veins to the heart so that it can be pumped to the tissues. Rt. Lt. At At Right Left ven ven Large veins Large arteries CAPACITANCE VESSELS EXCHANGE VESSELS RESISTANCE VESSELS Venules Capillaries Arterioles Vascular circuit • all 3 layers are present, but thinner than in •In varicose veins, arteries of corresponding size (external blood pools because diameter). -

Webinar Ch 8
Animal Anatomy and Physiology 1 Webinar Chapter 8 Cardiovascular System The Cardiovascular System Chapter 8 Pages 205-219 Textbook Learning Objectives Chapter 8 – Page 205 • List and describe the layers of the heart wall • List the chambers of the heart and describe the path of blood flow through the heart • Describe the structure and locations of the heart valves • Differentiate between systole and diastole • Describe the process of depolarization and repolarization of cardiac muscle cells • Describe the pathway of the electrical impulse generated by the SA node • List and describe the unique anatomical features of the fetal circulatory system • List the events responsible for the heart sounds heard on auscultation • List the factors that influence heart rate and cardiac output • Describe the relationship between cardiac output, heart rate, and stroke volume • Describe the structures of arteries, capillaries, and veins • List the major arteries and veins that travel from the heart to the systemic circulation • List the names and locations of veins commonly used for venipuncture in animals Cardiovascular System Heart Blood Vessels Blood YourDogsHeart.com http://www.yourdogsheart.com/ Your Cat’s Heart http://www.pethealthnetwork.com/cat-health/your- cat%E2%80%99s-heart-feline-heart-disease Great Human Heart A&P website http://www.nhlbi.nih.gov/health/health-topics/topics/hhw/ Internal Medicine The KEY Is Cellular Health Cellular Health • Healthy Cells = Health Animal Body • Diseased Cells = Diseased Animal Body • Too Many Diseased Cells -

Advanced P Wave Detection in Ecg Signals During Pathology
www.nature.com/scientificreports OPEN Advanced P Wave Detection in Ecg Signals During Pathology: Evaluation in Diferent Arrhythmia Contexts Lucie Maršánová 1*, Andrea Němcová1, Radovan Smíšek1,2, Martin Vítek1 & Lukáš Smital1 Reliable P wave detection is necessary for accurate and automatic electrocardiogram (ECG) analysis. Currently, methods for P wave detection in physiological conditions are well-described and efcient. However, methods for P wave detection during pathology are not generally found in the literature, or their performance is insufcient. This work introduces a novel method, based on a phasor transform, as well as innovative rules that improve P wave detection during pathology. These rules are based on the extraction of a heartbeats’ morphological features and knowledge of heart manifestation during both physiological and pathological conditions. To properly evaluate the performance of the proposed algorithm in pathological conditions, a standard database with a sufcient number of reference P wave positions is needed. However, such a database did not exist. Thus, ECG experts annotated 12 chosen pathological records from the MIT-BIH Arrhythmia Database. These annotations are publicly available via Physionet. The algorithm performance was also validated using physiological records from the MIT-BIH Arrhythmia and QT databases. The results for physiological signals were Se = 98.42% and PP = 99.98%, which is comparable to other methods. For pathological signals, the proposed method reached Se = 96.40% and PP = 85.84%, which greatly outperforms other methods. This improvement represents a huge step towards fully automated analysis systems being respected by ECG experts. These systems are necessary, particularly in the area of long-term monitoring. -

Heart Rate Variability
9 Heart Rate Variability I begin this chapter with a brief overview of relevant cardiac physiology. This section is intended for those who would like a refresher in cardiac physiology. If you are already well versed in physiology, this section may be something you glance through. Next, I introduce the concept of heart rate variability ( HRV ). In explaining the importance of HRV, I include a brief overview of supporting research. At the end of the chapter, I describe the main components of HRV and introduce a complete HRV training protocol that you can implement with your clients. Relevant Physiology The heart is a muscular organ that is responsible for pumping blood throughout the body with rhythmic contractions. The human heart has four chambers: two atria and two ventricles. The atria receive blood and the ventricles pump it out. Deoxygenated blood enters the right atrium, then goes into the right ventricle, and is then pumped out through the pulmonary arteries into the lungs. Reoxygenated blood returns from the lungs to the left atrium, then enters the left ventricle, and is then pumped out through the aorta to the rest of the body. Cardiac contractions are controlled by natural pacemakers: the sinoatrial node (SA) and the atrioventricular (AV) node. These nodes are self - excitable, meaning that they contract without any signal from the nervous system (they will continue contracting even if they are removed from the body). The SA node generates electri- cal impulses (action potentials; see Chapter 10 on surface electromyography (sEMG) The Clinical Handbook of Biofeedback: A Step-by-Step Guide for Training and Practice with Mindfulness, First Edition. -

Imaging and Modeling the Cardiovascular System
Imaging and modeling the cardiovascular system Elira Maksuti Doctoral Thesis KTH Royal Institute of Technology School of Technology and Health Department of Medical Engineering SE-141 57 Huddinge, Sweden Karolinska Institutet Department of Molecular Medicine and Surgery Clinical Physiology SE-171 76 Stockholm, Sweden TRITA-STH Report 2016:9 ISSN: 1653-3836 ISRN/KTH/STH/2016:9-SE ISBN: 978-91-7729-192-3 © Elira Maksuti, 2016 Printed in Sweden by Universitetsservice US-AB, Stockholm 2016 Preface This thesis is submitted to the KTH Royal Institute of Technology and Karolinska Institutet (KI) in partial fulfillment of the requirements for the Joint Doctoral Degree in Medical Technology. The work has mainly been performed at KTH School of Technology and Health in Huddinge, Sweden, and KI, Department of Clinical Physiology in Stockholm, Sweden, with Associate Professor Michael Broomé as main supervisor, Associate Professor Anna Bjällmark, Associate Professor Matilda Larsson and Associate Professor Martin Ugander as cosupervisors. Six months were spent at the Laboratory of Hemodynamics and Cardiovascular Technology at the École polytechnique fédérale de Lausanne (EPFL) in Lausanne, Switzerland, where Study III was performed, under the supervision of Professor Nikos Stergiopulos. The research projects were supported by grants from the Swedish Research Council (2012-2800, 2012-2795), VINNOVA VINNMER Marie Curie International Qualification Grant (2011-01365) and the Hans Werthén scholarship from The Royal Swedish Academy of Engineering Sciences (IVA). The thesis will be publicly defended at 10 am, December 9, 2016, in the T2 lecture hall, Hälsovägen 11C, Huddinge, Sweden. i ii Abstract Understanding cardiac pumping function is crucial to guiding diagnosis, predicting outcomes of interventions, and designing medical devices that interact with the cardiovascular system. -

In Physiology and Physics: Laws-Equations-Principles Professor Justiaan Swanevelder Head of Department UCT Dept of Anaesthesia & Perioperative Medicine
Part I Anaesthesia Refresher Course- 2017 01 University of Cape Town The “Big Five” in Physiology and Physics: Laws-Equations-Principles Professor Justiaan Swanevelder Head of Department UCT Dept of Anaesthesia & Perioperative Medicine Several physics equations are used to explain concepts of medical physics and human physiology, and are regularly applied in anaesthesia. Many of these concepts are being used on a daily basis in clinical practice without even thinking about the theory behind them. Philosophy gave birth to education and science! Most of the greatest scientists were also famous philosophers and started off with philosophical ideas. Several famous Philosopher-Physicists-Physiologists have their names connected to one or more “Law-Equation-Principle” in the medical field. Laws-Equations-Principles used every day & that should be understood in preparation for the Primary and Final FCA Exam, and for the rest of the anaesthetist/clinician’s working life, include: 1. Starling and compliance (cardiac function/mechanics, lung physiology, renal function) 2. La Place, Ohm (wall tension, preload, resistance, afterload, oxygen consumption) 3. Bernoulli (flow hydraulics, ultrasound-Doppler, pressure drop/flow across narrowing) 4. Fick, Shunt and Dead Space (cardiac, respiratory - diffusion, filters, oxygen supply and demand, monitoring) 5. Hagen-Poiseuille (flow through airways, tubes and tubing/vessels) Also important, but not covered in this lecture: 6. Boyle (Body plethysmography, “syringe, antibiotics, air and vial”) 7. Bohr (Haldane and Bohr Effects - O2-Hb Dissociation Curve, Dead Space Ventilation) 8. Henderson-Hasselbach (Blood gas interpretation) 9. Coanda (air/fluid flow pattern) 10. Venturi (gas entrailment, flow pattern) 11. Stewart-Hamilton (Cardiac Output by thermodilution) 12. -

Cardiac Output Venous Return
Circulatory Physiology As a Country Doc Episode 3 Cardiac Output and Venous Return Patrick Eggena, M.D. Novateur Medmedia, LLC. Circulatory Physiology As a Country Doc Episode 3 Cardiac Output and Venous Return Patrick Eggena, M.D. Novateur Medmedia, LLC. i Copyright This Episode is derived from: Course in Cardiovascular Physiology by Patrick Eggena, M.D. © Copyright Novateur Medmedia, LLC. April 13, 2012 The United States Copyright Registration Number: PAu3-662-048 Ordering Information via iBooks: ISBN 978-0-9663441-2-7 Circulatory Physiology as a Country Doc, Episode 3: Cardiac Output and Venous Return Contact Information: Novateur Medmedia, LLC 39 Terry Hill Road, Carmel, NY 10512 email: [email protected] Credits: Oil Paintings by Bonnie Eggena, PsD. Music by Alan Goodman from his CD Under the Bed, Cancoll Music, copyright 2005 (with permission). Illustrations, movies, text, and lectures by Patrick Eggena, M.D. Note: Knowledge in the basic and clinical sciences is constantly changing. The reader is advised to carefully consult the instruc- tions and informational material included in the package inserts of each drug or therapeutic agent before administration. The Country Doctor Series illustrates Physiological Principles and is not intended as a guide to Medical Therapeutics. Care has been taken to present correct information in this book, however, the author and publisher are not responsible for errors or omissions or for any consequence from application of the information in this work and make no warranty, expressed or implied, with re- spect to the contents of this publication or that its operation will be uninterrupted and error free on any particular recording de- vice.