Lawrence Berkeley National Laboratory Recent Work
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
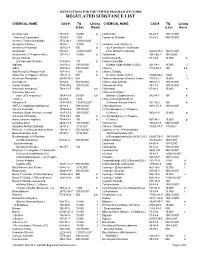
Regulated Substance List
INSTRUCTIONS FOR THE UNIFIED PROGRAM (UP) FORM REGULATED SUBSTANCE LIST CHEMICAL NAME CAS # TQ Listing CHEMICAL NAME CAS # TQ Listing (Lbs) Basis (Lbs) Basis Acetaldehyde 75-07-0 10,000 g Cantharidin 56-25-7 100/10,0001 * Acetone Cyanohydrin 75-86-5 1,000 Carbachol Chloride 51-83-2 500/10,0001 Acetone Thiosemicarbazide 1752-30-3 1,000/10,0001 Acetylene (Ethyne) 74-86-2 10,000 f Carbamic Acid, Methyl-,o- Acrolein (2-Propenal) 107-02-8 500 b (((2,4-Dimethyl-1,3-Dithiolan- Acrylamide 79-06-1 1,000/10,0001 2-YL) Methylene)Amino)- 26419-73-8 100/10,0001 Acrylonitrile (2- Propenenitrile) 107-13-1 10,000 b Carbofuran 1563-66-2 10/10,0001 Acrylyl Chloride Carbon Disulfide 75-15-0 10,000 b (2-Propenoyl Chloride) 814-68-6 100 b Carbon Oxysulfide Aldicarb 116-06-3 100/10,0001 (Carbon Oxide Sulfide (COS)) 463-58-1 10,000 f Aldrin 309-00-2 500/10,0001 Chlorine 7782-50-5 100 a,b Allyl Alcohol (2-Propen-1-ol) 107-18-6 1,000 b Chlorine Dioxide Allylamine (2-Propen-1-Amine) 107-11-9 500 b (Chlorine Oxide (ClO2)) 10049-04-4 1,000 c Aluminum Phosphide 20859-73-8 500 Chlorine Monoxide (Chlorine Oxide) 7791-21-1 10,000 f Aminopterin 54-62-6 500/10,0001 Chlormequat Chloride 999-81-5 100/10,0001 Amiton Oxalate 3734-97-2 100/10,0001 Chloroacetic Acid 79-11-8 100/10,0001 Ammonia, Anhydrous 2 7664-41-7 500 a,b Chloroform 67-66-3 10,000 b Ammonia, Aqueous Chloromethyl Ether (conc 20% or greater) 7664-41-7 20,000 a,b (Methane,Oxybis(chloro-) 542-88-1 100 b * Aniline 62-53-3 1,000 Chloromethyl Methyl Ether Antimycin A 1397-94-0 1,000/10,0001 (Chloromethoxymethane) -

Supporting Information For
Electronic Supplementary Material (ESI) for Chemical Communications This journal is © The Royal Society of Chemistry 2011 Supporting Information for: 2- Quadruple-CO3 Bridged Octanuclear Dysprosium(III) Compound Showing Single-Molecule Magnet Behaviour Experimental Section Synthetic procedures All chemicals were of reagent grade and were used without any further purification. Synthesis of pyrazine-2-carbohydrazide The pyrazine-2-carboxylic acid methyl ester was prepared by a literature procedure described elsewhere. A mixture of pyrazine-2-carboxylic acid methyl ester (1.38 g, 10 mmol) and hydrazine hydrate (85%, 15 ml) in methanol (20 ml) was refluxed overnight, at a temperature somewhat below 80°C. The resulting pale-yellow solution set aside 12 hrs. During this period, a colorless product, i.e. pyrazine-2-carbohydrazide, precipitated from the reaction mixture as a crystalline solid (yield = 0.84 g, 61%). Synthesis of (E)-N'-(2-hyborxy-3-methoxybenzylidene)pyrazine-2-carbohydrazide Pyrazine-2-carbohydrazide (2 mmol, 0.276 g) was suspended together with o-vanillin (2 mmol, 0.304 g) in methanol (20 ml), and the resulting mixture was stirred at the room temperature overnight. The pale yellow solid was collected by filtration (yield = 0.56 g, 83%). Elemental analysis (%) calcd for C13H12N4O3: C, 57.35, H, 4.44, N, 20.58: found C, 57.64, H, 4.59, N, 20.39. IR (KBr, cm-1): 3415(w), 3258(w), 1677(vs), 1610(s), 1579(m). 1530(s), 1464(s), 1363(m), 1255(vs), 1153(s), 1051(w), 1021(s), 986(w), 906(m), 938(w), 736(s), 596(m), 498(w). Synthesis of the complex 1 The solution of DyCl3⋅6H2O (56.5 mg, 0.15 mmol) and the H2L (40.5 mg, 0.15 mmol) in 15 ml CH3OH/CH2Cl2 (1:2 v/v) was stirred with triethylamine (0.4 mmol) for 6h. -
Densifying Metal Hydrides with High Temperature and Pressure
3,784,682 United States Patent Office Patented Jan. 8, 1974 feet the true density. That is, by this method only theo- 3,784,682 retical or near theoretical densities can be obtained by DENSIFYING METAL HYDRIDES WITH HIGH making the material quite free from porosity (p. 354). TEMPERATURE AND PRESSURE The true density remains the same. Leonard M. NiebylsM, Birmingham, Mich., assignor to Ethyl Corporation, Richmond, Va. SUMMARY OF THE INVENTION No Drawing. Continuation-in-part of abandoned applica- tion Ser. No. 392,370, Aug. 24, 1964. This application The process of this invention provides a practical Apr. 9,1968, Ser. No. 721,135 method of increasing the true density of hydrides of Int. CI. COlb 6/00, 6/06 metals of Groups II-A, II-B, III-A and III-B of the U.S. CI. 423—645 8 Claims Periodic Table. More specifically, true densities of said 10 metal hydrides may be substantially increased by subject- ing a hydride to superatmospheric pressures at or above ABSTRACT OF THE DISCLOSURE fusion temperatures. When beryllium hydride is subjected A method of increasing the density of a hydride of a to this process, a material having a density of at least metal of Groups II-A, II-B, III-A and III-B of the 0.69 g./cc. is obtained. It may or may not be crystalline. Periodic Table which comprises subjecting a hydride to 15 a pressure of from about 50,000 p.s.i. to about 900,000 DESCRIPTION OF THE PREFERRED p.s.i. at or above the fusion temperature of the hydride; EMBODIMENT i.e., between about 65° C. -

Thermodynamic Hydricity of Small Borane Clusters and Polyhedral Closo-Boranes
molecules Article Thermodynamic Hydricity of Small Borane Clusters y and Polyhedral closo-Boranes Igor E. Golub 1,* , Oleg A. Filippov 1 , Vasilisa A. Kulikova 1,2, Natalia V. Belkova 1 , Lina M. Epstein 1 and Elena S. Shubina 1,* 1 A. N. Nesmeyanov Institute of Organoelement Compounds and Russian Academy of Sciences (INEOS RAS), 28 Vavilova St, 119991 Moscow, Russia; [email protected] (O.A.F.); [email protected] (V.A.K.); [email protected] (N.V.B.); [email protected] (L.M.E.) 2 Faculty of Chemistry, M.V. Lomonosov Moscow State University, 1/3 Leninskiye Gory, 119991 Moscow, Russia * Correspondence: [email protected] (I.E.G.); [email protected] (E.S.S.) Dedicated to Professor Bohumil Štibr (1940-2020), who unfortunately passed away before he could reach the y age of 80, in the recognition of his outstanding contributions to boron chemistry. Academic Editors: Igor B. Sivaev, Narayan S. Hosmane and Bohumír Gr˝uner Received: 6 June 2020; Accepted: 23 June 2020; Published: 25 June 2020 MeCN Abstract: Thermodynamic hydricity (HDA ) determined as Gibbs free energy (DG◦[H]−) of the H− detachment reaction in acetonitrile (MeCN) was assessed for 144 small borane clusters (up 2 to 5 boron atoms), polyhedral closo-boranes dianions [BnHn] −, and their lithium salts Li2[BnHn] (n = 5–17) by DFT method [M06/6-311++G(d,p)] taking into account non-specific solvent effect (SMD MeCN model). Thermodynamic hydricity values of diborane B2H6 (HDA = 82.1 kcal/mol) and its 2 MeCN dianion [B2H6] − (HDA = 40.9 kcal/mol for Li2[B2H6]) can be selected as border points for the range of borane clusters’ reactivity. -

Diborane (B2H6) 2 Heavier Boron Hydrides in the Form of Volatile Liquids Such As
Even in the absence of contamination, diborane is so reactive that it will slowly decompose by itself, forming H gas and Diborane (B2H6) 2 heavier boron hydrides in the form of volatile liquids such as Storage and Delivery B5H9 and sublimable solids such as B10H14. These heavier hydrides are delivered with the gas and can cause Air Products supplies a wide range of gases and chemicals contamination of the process and fouling of the delivery system that are used for integrated circuit, flat panel display and components. Figure 1 shows the internals of the first stage of photovoltaic device manufacturing. Understanding the proper a two-stage gas regulator that was fouled by B10H14 deposits. storage and handling requirements of these specialty materials is essential to obtain consistent results and to ensure the highest level of process safety. In this technical bulletin, we will provide the user with some of the knowledge required for its effective storage and use. Figure 1: Picture of the first stage of diborane mixture regulator Diborane (formula B2H6) is a colorless, spontaneously fouled with solid decomposition products. flammable (pyrophoric) gas. It burns rapidly in air and reacts vigorously with water with the evolution of heat. The gas has The gas-phase decomposition rate of diborane was shown to a distinct, noxious odor and is extremely toxic by inhalation or increase with concentration and with temperature. Pure by skin exposure. Because of these hazards, it is essential diborane is so reactive in its pure state that it may only be that all gas handling systems used for diborane be tightly stored or transported if surrounded with dry ice to prevent rapid sealed and carefully leak checked. -

Germane Facts About Germanium Sesquioxide: I. Chemistry and Anticancer Properties
THE JOURNAL OF ALTERNATIVE AND COMPLEMENTARY MEDICINE Volume 10, Number 2, 2004, pp. 337–344 ©Mary Ann Liebert, Inc. Germane Facts About Germanium Sesquioxide: I. Chemistry and Anticancer Properties BONNIEJ. KAPLAN, Ph.D., 1 W. WESLEYPARISH, Ph.D., 2 G. MERRILLANDRUS, Ph.D., 2 J. STEVENA. SIMPSON, Ph.D., M.D., 3 and CATHERINEJ. FIELD, Ph.D., R.D. 4 ABSTRACT This paper reviews the history, chemistry, safety, toxicity, and anticancer effects of the organogermanium compound bis (2-carboxyethylgermanium) sesquioxide (CEGS). A companion review follows, discussing the inaccuracies in the scientific record that have prematurely terminated research on clinical uses of CEGS. CEGS is a unique organogermanium compound first made by Mironov and coworkers in Russia and, shortly there- after, popularized by Asai and his colleagues in Japan. Low concentrations of germanium occur in nearly all soils, plants and animal life; natural occurrence of the CEGS form is postulated but not yet demonstrated. The literature demonstrating its anticancer effect is particularly strong: CEGS induces interferon- g (IFN-g), en- hances natural killer cell activity, and inhibits tumor and metastatic growth—effects often detectable after a single oral dose. In addition, oral consumption of CEGS is readily assimilated and rapidly cleared from the body without evidence of toxicity. Given these findings, the absence of human clinical trials of CEGS is un- expected. Possible explanations of why the convincing findings from animal research have not been used to support clinical trials are discussed. Clinical trials on CEGS are recommended. INTRODUCTION bispropionic acid; 3-oxygermylpropionic acid polymer; poly- trans-(2-carboxyethyl) germasesquioxane); proxigerma- n general, dietary supplements are an underinvestigated nium; repagermanium; and Serocion. -

Hydrous Hydrazine Decomposition for Hydrogen Production Using of Ir/Ceo2: Effect of Reaction Parameters on the Activity
nanomaterials Article Hydrous Hydrazine Decomposition for Hydrogen Production Using of Ir/CeO2: Effect of Reaction Parameters on the Activity Davide Motta 1, Ilaria Barlocco 2 , Silvio Bellomi 2, Alberto Villa 2,* and Nikolaos Dimitratos 3,* 1 Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff CF10 3AT, UK; [email protected] 2 Dipartimento di Chimica, Università degli Studi di Milano, 20133 Milano, Italy; [email protected] (I.B.); [email protected] (S.B.) 3 Dipartimento di Chimica Industriale e dei Materiali, Alma Mater Studiorum Università di Bologna, 40136 Bologna, Italy * Correspondence: [email protected] (A.V.); [email protected] (N.D.) Abstract: In the present work, an Ir/CeO2 catalyst was prepared by the deposition–precipitation method and tested in the decomposition of hydrazine hydrate to hydrogen, which is very important in the development of hydrogen storage materials for fuel cells. The catalyst was characterised using different techniques, i.e., X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM), scanning electron microscopy (SEM) equipped with X-ray detector (EDX) and inductively coupled plasma—mass spectroscopy (ICP-MS). The effect of reaction conditions on the activity and selectivity of the material was evaluated in this study, modifying parameters such as temperature, the mass of the catalyst, stirring speed and concentration of base in order to find the optimal conditions of reaction, which allow performing the test in a kinetically limited regime. Citation: Motta, D.; Barlocco, I.; Bellomi, S.; Villa, A.; Dimitratos, N. Keywords: iridium; cerium oxide; hydrous hydrazine; hydrogen production Hydrous Hydrazine Decomposition for Hydrogen Production Using of Ir/CeO2: Effect of Reaction Parameters on the Activity. -

276262828.Pdf
FUNDAMENTAL STUDIES OF CATALYTIC DEHYDROGENATION ON ALUMINA-SUPPORTED SIZE-SELECTED PLATINUM CLUSTER MODEL CATALYSTS by Eric Thomas Baxter A dissertation submitted to the faculty of The University of Utah in partial fulfillment of the requirements for the degree of Doctor of Philosophy Department of Chemistry The University of Utah May 2018 Copyright © Eric Thomas Baxter 2018 All Rights Reserved T h e U n iversity of Utah Graduate School STATEMENT OF DISSERTATION APPROVAL The dissertation of Eric Thomas Baxter has been approved by the following supervisory committee members: Scott L. Anderson , Chair 11/13/2017 Date Approved Peter B. Armentrout , Member 11/13/2017 Date Approved Marc D. Porter , Member 11/13/2017 Date Approved Ilya Zharov , Member 11/13/2017 Date Approved Sivaraman Guruswamy , Member 11/13/2017 Date Approved and by Cynthia J. Burrows , Chair/Dean of the Department/College/School of Chemistry and by David B. Kieda, Dean of The Graduate School. ABSTRACT The research presented in this dissertation focuses on the use of platinum-based catalysts to enhance endothermic fuel cooling. Chapter 1 gives a brief introduction to the motivation for this work. Chapter 2 presents fundamental studies on the catalytic dehydrogenation of ethylene by size-selected Ptn (n = 4, 7, 8) clusters deposited onto thin film alumina supports. The model catalysts were probed by a combination of experimental and theoretical techniques including; temperature-programmed desorption and reaction (TPD/R), low energy ion scattering spectroscopy (ISS), X-ray photoelectron spectroscopy (XPS), plane wave density-functional theory (PW-DFT), and statistical mechanical theory. It is shown that the Pt clusters dehydrogenated approximately half of the initially adsorbed ethylene, leading to deactivation of the catalyst via (coking) carbon deposition. -

Theoretical Study of the Bis-Silylation Reaction of Ethylene Catalyzed by Titanium Dichloride Yuri Alexeev Iowa State University
Chemistry Publications Chemistry 8-2003 Theoretical Study of the Bis-Silylation Reaction of Ethylene Catalyzed by Titanium Dichloride Yuri Alexeev Iowa State University Mark S. Gordon Iowa State University, [email protected] Follow this and additional works at: http://lib.dr.iastate.edu/chem_pubs Part of the Chemistry Commons The ompc lete bibliographic information for this item can be found at http://lib.dr.iastate.edu/ chem_pubs/419. For information on how to cite this item, please visit http://lib.dr.iastate.edu/ howtocite.html. This Article is brought to you for free and open access by the Chemistry at Iowa State University Digital Repository. It has been accepted for inclusion in Chemistry Publications by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Theoretical Study of the Bis-Silylation Reaction of Ethylene Catalyzed by Titanium Dichloride Abstract Titanium dichloride was investigated as a potential catalyst for the bis-silylation reaction of ethylene with hexachlorodisilane. Ab initio electronic structure calculations at the restricted Hartree−Fock (RHF), density functional (DFT), second-order perturbation (MP2), and couple cluster (CCSD) levels of theory were used to find optimized structures, saddle points, and minimum-energy paths that connect them. The er action was found to have a net zero barrier at the DFT, MP2, and CCSD levels of theory. Dynamic correlation is found to be important for this reaction. Disciplines Chemistry Comments Reprinted (adapted) with permission from Organometallics 22 (2003): 4111, doi:10.1021/om0303350. Copyright 2014 American Chemical Society. This article is available at Iowa State University Digital Repository: http://lib.dr.iastate.edu/chem_pubs/419 Organometallics 2003, 22, 4111-4117 4111 Theoretical Study of the Bis-Silylation Reaction of Ethylene Catalyzed by Titanium Dichloride Yuri Alexeev and Mark S. -

Germane 99.99+%
GEG5001 - GERMANE 99.99+% GERMANE 99.99+% Safety Data Sheet GEG5001 Date of issue: 01/05/2015 Version: 1.0 SECTION 1: Identification of the substance/mixture and of the company/undertaking 1.1. Product identifier Product form : Substance Physical state : Gas Substance name : GERMANE 99.99+% Product code : GEG5001 Formula : GeH4 Synonyms : MONOGERMANE; GERMANIUM HYDRIDE; GERMANIUM TETRAHYDRIDE Chemical family : GERMANE 1.2. Relevant identified uses of the substance or mixture and uses advised against Use of the substance/mixture : Chemical intermediate For research and industrial use only 1.3. Details of the supplier of the safety data sheet GELEST, INC. 11 East Steel Road Morrisville, PA 19067 USA T 215-547-1015 - F 215-547-2484 - (M-F): 8:00 AM - 5:30 PM EST [email protected] - www.gelest.com 1.4. Emergency telephone number Emergency number : CHEMTREC: 1-800-424-9300 (USA); +1 703-527-3887 (International) SECTION 2: Hazards identification 2.1. Classification of the substance or mixture Classification (GHS-US) Flam. Gas 1 H220 Liquefied gas H280 Acute Tox. 2 (Inhalation:gas) H330 Eye Irrit. 2A H319 STOT SE 3 H335 Full text of H-phrases: see section 16 2.2. Label elements GHS-US labeling Hazard pictograms (GHS-US) : GHS02 GHS04 GHS06 GHS07 Signal word (GHS-US) : Danger Hazard statements (GHS-US) : H220 - Extremely flammable gas H280 - Contains gas under pressure; may explode if heated H319 - Causes serious eye irritation H330 - Fatal if inhaled H335 - May cause respiratory irritation Precautionary statements (GHS-US) : P284 - [In case of inadequate ventilation] wear respiratory protection P280 - Wear protective gloves/protective clothing/eye protection/face protection P260 - Do not breathe gas P264 - Wash hands thoroughly after handling P310 - Immediately call a doctor P210 - Keep away from heat, open flames, sparks. -

Solid-State Structures of the Covalent Hydrides Germane and Stannane
Edinburgh Research Explorer Solid-state structures of the covalent hydrides germane and stannane Citation for published version: Maley, IJ, Brown, DH, Ibberson, RM & Pulham, CR 2008, 'Solid-state structures of the covalent hydrides germane and stannane', Acta Crystallographica Section B - Structural Science, vol. 64, no. Pt 3, pp. 312-7. https://doi.org/10.1107/S0108768108010379 Digital Object Identifier (DOI): 10.1107/S0108768108010379 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: Acta Crystallographica Section B - Structural Science Publisher Rights Statement: Copyright © 2008 International Union of Crystallography; all rights reserved. General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 02. Oct. 2021 electronic reprint Acta Crystallographica Section B Structural Science, Crystal Engineering and Materials ISSN 2052-5192 Solid-state structures of the covalent hydrides germane and stannane Iain J. Maley, Daniel H. Brown, Richard M. Ibberson and Colin R. Pulham Acta Cryst. (2008). B64, 312–317 Copyright c International Union of Crystallography Author(s) of this paper may load this reprint on their own web site or institutional repository provided that this cover page is retained. -

Hazardous Material Inventory Statement
City of Brooklyn Park FIRE DEPARTMENT 5200 - 85th Avenue North Brooklyn Park MN 55443 Phone: (763)493-8020 Fax: (763) 493-8391 Hazardous Materials Inventory Statement Users Guide A separate inventory statement shall be provided for each building. An amended inventory statement shall be provided within 30 days of the storage of any hazardous materials or plastics that changes or adds a hazard class or which is sufficient in quantity to cause an increase in the quantity which exceeds 5 percent for any hazard class. The hazardous materials inventory statement shall list by hazard class categories. Each grouping shall provide the following information for each hazardous material listed for that group including a total quantity for each group of hazard class. 1. Hazard class. (See attached Hazardous Materials Categories Listing) 2. Common or trade name. 3. Chemical Abstract Service Number (CAS number) found in 29 Code of Federal Regulations (C.F.R.). 4. Whether the material is pure or a mixture, and whether the material is a solid, liquid or gas 5. Maximum aggregate quantity stored at any one time. 6. Maximum aggregate quantity In-Use (Open to atmosphere) at any one time. 7. Maximum aggregate quantity In-Use (Closed to atmosphere) at any one time. 8. Storage conditions related to the storage type, high-pile, encapsulated, non-encapsulated. Attached is a listing of categories that all materials need to be organized to. Definitions of these categories are also attached for your use. At the end of this packet are blank forms for completing this project. For questions regarding Hazardous Materials Inventory Statement contact the Fire Department at 763-493-8020.