021879Orig1s000
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(12) United States Patent (10) Patent No.: US 6,844,355 B2 Somberg Et Al
USOO684.4355B2 (12) United States Patent (10) Patent No.: US 6,844,355 B2 Somberg et al. (45) Date of Patent: Jan. 18, 2005 (54) OPTICALLY ACTIVE ISOMERS OF QUININE Catterall, (1992) Physiol. Rev. 72(supp):S15-S48. AND QUINDINE AND THEIR RESPECTIVE Chan et al., (1991) J. Chromatogr. 571:291-297. BIOLOGICALACTION Chen et al., (1998) Nature 392:293–296. (75) Inventors: John C. Somberg, Lake Forest, IL Coplen et al., (1991) Circulation 84:527. (US); Vasant Ranade, Libertyville, IL Drabowicz et al., (1984) “Chemical Abstracts” Phosphorus (US) Sulfur 16:2676–270 (XP002165239). Engler et al., (1985) Helv. Chim. Acta. 68:789–800. (73) Assignee: Academic Pharmaceuticals, Inc., Lake Ficker et al., (1998) Circ. Res. 82:386–395. Bluff, IL (US) Gellens et al., (1992) Proc. Natl. Acad. Sci. 89:554–558. (*) Notice: Subject to any disclaimer, the term of this Gutzwiller et al., (1973) “Chemical Abstracts” Helv. Chim. patent is extended or adjusted under 35 Acta. 79:1494–1503 (XP002165238). U.S.C. 154(b) by 0 days. Hartmann et al. (1994) Circ. Res. 75:114-122. Karle I.L. et al., (1981) Proc. Natl. Acad. Sci. 78:5938–5941. (21) Appl. No.: 10/168,919 Karle, J.M. (1997) Antimicrob. Agents Chemother. (22) PCT Filed: Dec. 22, 2000 41:791-794. Kiehn et al. (1996) Circulation 94:2572–2579. (86) PCT No.: PCT/US00/352.13 Krafte et al., (1994) Europ. J. Pharma. 266:245-254. S371 (c)(1), Li et al. (1996) Circ. Res. 78:689-696. (2), (4) Date: Oct. 30, 2002 Sanguinetti et al., (1990) J. -

Dietary Supplements Compendium Volume 1
2015 Dietary Supplements Compendium DSC Volume 1 General Notices and Requirements USP–NF General Chapters USP–NF Dietary Supplement Monographs USP–NF Excipient Monographs FCC General Provisions FCC Monographs FCC Identity Standards FCC Appendices Reagents, Indicators, and Solutions Reference Tables DSC217M_DSCVol1_Title_2015-01_V3.indd 1 2/2/15 12:18 PM 2 Notice and Warning Concerning U.S. Patent or Trademark Rights The inclusion in the USP Dietary Supplements Compendium of a monograph on any dietary supplement in respect to which patent or trademark rights may exist shall not be deemed, and is not intended as, a grant of, or authority to exercise, any right or privilege protected by such patent or trademark. All such rights and privileges are vested in the patent or trademark owner, and no other person may exercise the same without express permission, authority, or license secured from such patent or trademark owner. Concerning Use of the USP Dietary Supplements Compendium Attention is called to the fact that USP Dietary Supplements Compendium text is fully copyrighted. Authors and others wishing to use portions of the text should request permission to do so from the Legal Department of the United States Pharmacopeial Convention. Copyright © 2015 The United States Pharmacopeial Convention ISBN: 978-1-936424-41-2 12601 Twinbrook Parkway, Rockville, MD 20852 All rights reserved. DSC Contents iii Contents USP Dietary Supplements Compendium Volume 1 Volume 2 Members . v. Preface . v Mission and Preface . 1 Dietary Supplements Admission Evaluations . 1. General Notices and Requirements . 9 USP Dietary Supplement Verification Program . .205 USP–NF General Chapters . 25 Dietary Supplements Regulatory USP–NF Dietary Supplement Monographs . -

Inherited Heart Conditions Sudden Arrhythmic Death Syndrome
Inherited heart conditions Sudden arrhythmic death syndrome In association with Contents Introduction 04 Understanding Your Heart The normal heart 07 Sudden cardiac death and Sudden Arrhythmic Death Syndrome (SADS) 09 What happens after an unexpected sudden death? 10 Conditions that cause SADS What causes SADS? 13 Long QT Syndrome (LQTS) 14 Brugada Syndrome 18 CPVT (catecholaminergic polymorphic ventricular tachycardia) 21 PCCD (progressive cardiac conduction defect) 22 IVF (idiopathic ventricular fibrillation) 23 Sodium channel disease 23 Structural heart disease 23 Mitral valve prolapse 24 SADS and your family Implications of a SADS death for close blood Relatives of the person who has died 27 Assessment at a clinic for inherited cardiac conditions 28 Everyday Life General lifestyle advice 37 Author: Drugs to avoid 38 Dr Elijah R Behr MD Looking Forward Senior Lecturer and Honorary Consultant Electrophysiologist, Cardiology and Cardiological Sciences, St George’s Hospital and University of London The future 49 Published by Cardiac Risk in the Young (CRY) and the British Heart Foundation. Technical terms 51 This booklet is not a substitute for the advice your doctor or cardiologist (heart For more information 57 specialist) may give you based on his or her knowledge of your condition, but it should help you to understand what they tell you. Index 58 Introduction Title of chapter Introduction Understanding your heart You may be reading this booklet because a relative of yours – perhaps a member of your own family – has died suddenly and unexpectedly. This is not only a tragedy for the person and all your family, but a great loss for society too. -
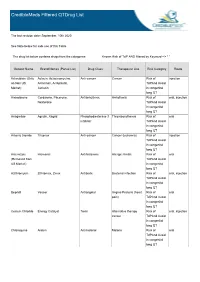
Crediblemeds Filtered Qtdrug List
CredibleMeds Filtered QTDrug List The last revision date: September, 10th 2020 See Note below for safe use of this Table The drug list below contains drugs from the categories: Known Risk of TdP AND filtered by Keyword --> " " Generic Name Brand Names (Partial List) Drug Class Therapeutic Use Risk Category Route Aclarubicin (Only Aclacin, Aclacinomycine, Anti-cancer Cancer Risk of injection on Non US Aclacinon, Aclaplastin, TdPAnd Avoid Market) Jaclacin in congenital long QT Amiodarone Cordarone, Pacerone, Antiarrhythmic Arrhythmia Risk of oral, injection Nexterone TdPAnd Avoid in congenital long QT Anagrelide Agrylin, Xagrid Phosphodiesterase 3 Thrombocythemia Risk of oral inhibitor TdPAnd Avoid in congenital long QT Arsenic trioxide Trisenox Anti-cancer Cancer (leukemia) Risk of injection TdPAnd Avoid in congenital long QT Astemizole Hismanal Antihistamine Allergic rhinitis Risk of oral (Removed from TdPAnd Avoid US Market) in congenital long QT Azithromycin Zithromax, Zmax Antibiotic Bacterial infection Risk of oral, injection TdPAnd Avoid in congenital long QT Bepridil Vascor Antianginal Angina Pectoris (heart Risk of oral pain) TdPAnd Avoid in congenital long QT Cesium Chloride Energy Catalyst Toxin Alternative therapy Risk of oral, injection cancer TdPAnd Avoid in congenital long QT Chloroquine Aralen Antimalarial Malaria Risk of oral TdPAnd Avoid in congenital long QT Generic Name Brand Names (Partial List) Drug Class Therapeutic Use Risk Category Route Chlorpromazine Thorazine, Largactil, Antipsychotic / Nausea, Schizophrenia, -
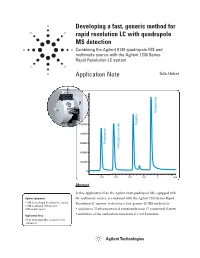
Developing a Fast, Generic Method for Rapid Resolution LC with Quadrupole
Developing a fast, generic method for rapid resolution LC with quadrupole MS detection Combining the Agilent 6140 quadrupole MS and multimode source with the Agilent 1200 Series Rapid Resolution LC system Application Note Udo Huber 1600000 1400000 Disopyramide 1200000 1000000 Quinidine 800000 600000 Procainamide N-Acetylprocainamide 400000 200000 0 0.2 0.4 0.6 0.8 1 min Abstract In this Application Note the Agilent 6140 quadrupole MS, equipped with Agilent Equipment the multimode source, is combined with the Agilent 1200 Series Rapid • 1200 Series Rapid Resolution LC system Resolution LC system, to develop a fast, generic LC/MS method for: • 6140 quadrupole MS detector • Multimode Source • analysis of 35 pharmaceutical compounds from 17 compound classes • reduction of the method run time from 2.1 to 0.8 minutes Application Area • High throughput MS analysis in drug discovery Introduction Experimental • One resistive capillary (Agilent part number G1960-80060) With the introduction of the Agilent Equipment • One narrow-bore capillary 1200 Series Rapid Resolution LC The Agilent 1200 Series Rapid 1 (Agilent part number G1960- system , it is now possible to run Resolution LC/MS system con- 20310). sub-2-micron particle columns at tained the following modules: high flow rates and achieve run • Agilent 1200 Series binary pump times of less than 1 – 2 minutes. SL Results and discussion With increased temperatures, 4.6 • Agilent 1200 Series high-perfor- mm i.d. columns can be operated at mance autosampler SL Moving towards a generic method flow rates of 5 mL/min but this flow • Agilent 1200 Series thermostat- Generic methods for liquid chro- rate is too high for electrospray ted column compartment SL matography with UV detection often (ESI) or atmospheric pressure • Agilent 1200 Series diode array use trifluoro acetic acid (TFA) as chemical ionization (APCI). -

PAUL, Balbir Singh, 1925- a STUDY on the PHARMACODYNAMICS of DIHYDROQUINIDINE in CANINE
This dissertation has been microfilmed exactly as received 67-16,326 PAUL, Balbir Singh, 1925- A STUDY ON THE PHARMACODYNAMICS OF DIHYDROQUINIDINE IN CANINE. The Ohio State University, Ph.D,, 1967 Pharmacology University Microfilms, Inc., Ann Arbor, Michigan A STUDY ON THE PHARMACODYNAMICS OF DIHYDROQUINIDINE IN CANINE DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Balbir Singh Paul, B.V.Sc., M.V.Sc. ******* The Ohio State University 1967 Approved by Adviser Department of Veterinary Physiology and Pharmacology ACKNOWLEDGMENTS I am highly indebted to Dr. Thomas E. Powers, Professor in the department of Veterinary Physiology and Pharmacology for his excellent guidance, encouragement and the sincere interest shown by him throughout the entire course of my graduate program-and research. My sincere thanks are due to Drs. S. Dutta and B. Marks for cooperation and affording facilities and guid ance during the course of my research. Special thanks are also due to Drs. C. R. Smith, R. W. Redding and R. S. Ray for giving their valid criticism and guidance during the course of my dissertation writing. The numerous illustrations were drawn by Mr. B. Kramer and Miss Diana Walker, medical artists. The statis tical computations in part, for this study, were done in the statistical laboratory at The Ohio State University. Technical assistance was given by Mrs. Dassie Sprecher, Messrs. Thomas Heading and Robert Slobody. I am pleased to have been able to fulfill the long desire of my late father. Dr. Ram Singh, in acquiring my graduate education. -

Quinidine Assay Preservatives, and Stabilizers Precautions • for in Vitro Diagnostic Use
4 REAGENTS Reagents contain the following substances: Mouse monoclonal antibodies reactive to quinidine (1.35 µg/mL), glucose-6-phosphate (22 mM), nicotinamide adenine dinucleotide (18 mM), quinidine labeled with glucose-6-phosphate dehydrogenase (0.41 U/mL), 0.1% sodium azide, Tris buffer, Quinidine Assay preservatives, and stabilizers Precautions • For in vitro diagnostic use. July 2012 4Q052.2D_C • Contains nonsterile mouse monoclonal antibodies. • Assay components contain sodium azide, which may react with lead and copper plumbing to See shaded sections: form highly explosive metal azides. If waste is discarded down the drain, flush it with a large Updated information from August 2010 edition. volume of water to prevent azide buildup. • Do not use the kit after the expiration date. • This kit contains streptomycin sulfate. Please dispose of appropriately. • Turbid or yellow reagents may indicate contamination or degradation and must be discarded. Safety data sheets (MSDS/SDS) available on www.siemens.com/diagnostics Catalog Quantity/ Preparation of Reagents Number Product Description Volume The Emit® 2000 Quinidine Assay reagents are provided ready to use; no preparation is OSR4Q229 Emit® 2000 Quinidine Assay necessary. OSR4Q518 R1 (Antibody/Substrate Reagent 1) 2 x 23 mL Storage of Assay Components OSR4Q548 R2 (Enzyme Reagent 2) 2 x 13 mL • Improper storage of reagents can affect assay performance. • When not in use, store reagents upright at 2–8°C and with screw caps tightly closed. 4Q109UL Emit® 2000 Quinidine Calibrators* 1 x 5 mL†, 5 x 2 mL • Unopened reagents are stable until the expiration date printed on the label if stored upright at 2–8°C. -

Application Note 4675
Application Note 135 Antiarrhythmic Drugs on Short Columns, Using Simple Mobile Phase Adjustments Quick analyses of antiarrhythmic drugs are necessary for Figure A. Antiarrhythmic Drugs Resolved within clinical laboratories to process large numbers of samples. Classes through Simple Mobile Phase Adjustments By using the 5cm SUPELCOSIL LC-8-DB column, with only Column: SUPELCOSIL LC-8-DB (58344), 5cm x 4.6mm, two stock mobile phase solutions, clinicians can sequen- 5µm packing, with Supelguard guard column (59553), 2cm x 4.6mm, 5µm packing tially analyze five classes of antiarrhythmic drugs and Col. Temp.: 30°C Mobile Phase: (A1) 5% acetonitrile:95% 0.02M H PO achieve accurate chromatography. 3 4 (pH to 3.0 with KOH) + 0.02% triethylamine, pH 3.3 (A2) 10% acetonitrile:90% 0.02M H3PO4 (A3) 15% acetonitrile:85% 0.02M H PO Key Words: 3 4 (A4, A5) 20% acetonitrile:80% 0.02M H3PO4 l antiarrhythmic drugs l HPLC Flow Rate: 2.0mL/min Det.: (A1) 280nm UV, 0.2 AUFS l SUPELCOSIL HPLC columns l solid phase extraction (A2) 215nm UV, 0.4 AUFS (A3) 254nm UV, 0.02 AUFS (A4) 215nm UV, 0.3 AUFS Many times, physicians prescribe drugs that are effective in small (A5) 215nm UV, 0.02 AUFS doses at low blood levels and have relatively high toxicity (1). Sample: 10µL mobile phase, on-column quantities of drugs listed on figure The blood level of one or more active metabolites must often be A1– Procainamide A2 – Lidocaine determined simultaneously with the administered drug. This Glycine xylidide Procainamide (33ng) makes imperative the need for accurate, rapid drug monitoring (33ng) procedures. -

Pharmacological Drugs Inducing Ototoxicity, Vestibular Symptoms and Tinnitus: a Reasoned and Updated Guide
European Review for Medical and Pharmacological Sciences 2011; 15: 601-636 Pharmacological drugs inducing ototoxicity, vestibular symptoms and tinnitus: a reasoned and updated guide G. CIANFRONE1, D. PENTANGELO1, F. CIANFRONE2, F. MAZZEI1, R. TURCHETTA1, M.P. ORLANDO1, G. ALTISSIMI1 1Department of Otolaryngology, Audiology and Phoniatrics, “Umberto I” University Hospital, Sapienza University, Rome (Italy); 2Institute of Otorhinolaryngology, School of Medicine, Catholic University of the Sacred Heart, Rome (Italy) Abstract. – The present work on drug-in- Introduction duced ototoxicity, tinnitus and vertigo repre- sents the update and revision of a previous The panorama of the pharmacological origin guide to adverse drug reactions for italian physi- iatrogenic noxae able to induce either harmful cians (2005). The panorama of drug-induced side effects causing ototoxicity or symptoms ototoxic effects or just a symptomatology like tin- such as tinnitus or dizziness and vertigo has en- nitus or balance disturbances, without any harm- larged in recent years, thanks to a better knowl- ful consequence, has widened in the last few edge and a more specific attention of pharma- years. The reason for this is the progress of scien- ceutical firms and drug-control institutions. In tific knowledge, the increased awareness of the daily clinical practice, there is a need for the pharmaceutical companies and of the institutions, family physician and the ENT specialist or audi- ologist (also in consideration of the possible which supervise pharmaceutical production. medico-legal implications) to focus the attention Only through continuous updating and experi- on the possible risk of otological side effects. ence sharing it’s possible to offer patients the This would allow a clinical risk-benefit evalua- certainty of receiving the treatment that is appro- tion, weighing the possible clinical advantage in priate, safe and effective and based upon the their field of competence against possible oto- most credited clinical studies. -

Simultaneous High Performance Liquid Chromatographic Determination Of
Rochester Institute of Technology RIT Scholar Works Theses Thesis/Dissertation Collections 5-1-1981 Simultaneous high performance liquid chromatographic determination of procainamide, N-acetylprocainamide, disopyramide, mono-N- dealkyldisopyramide, quinidine, and propranolol in serum James F. Wesley Follow this and additional works at: http://scholarworks.rit.edu/theses Recommended Citation Wesley, James F., "Simultaneous high performance liquid chromatographic determination of procainamide, N-acetylprocainamide, disopyramide, mono-N-dealkyldisopyramide, quinidine, and propranolol in serum" (1981). Thesis. Rochester Institute of Technology. Accessed from This Thesis is brought to you for free and open access by the Thesis/Dissertation Collections at RIT Scholar Works. It has been accepted for inclusion in Theses by an authorized administrator of RIT Scholar Works. For more information, please contact [email protected]. SIMULTANEOUS HIGH PERFORMANCE LIQUID CHROMATOGRAPHIC DETERMINATION OF PROCAINAMIDE, N-ACETYLPROCAINAMIDE, DISOPYRAMIDE, MONO-N-DEALKYLDISOPYRAMIDE, QUINIDINE, AND PROPRANOLOL IN SERUM James F. Wesley May 1981 Submitted as part of the requirements for a Master of Science Degree in Clinical Chemistry at Rochester Institute of Technology Rochester, New York 14623 Department of Clinical Sciences PERMISSION FOR REPRODUCTION Simultaneous high performance liquid chromatographic determination of procainamide, N-acetyl procainamide, disopyramide, mono-N-dealkyldiso- pyramide, quinidine, and propranolol in serum. I, James F. -

Quinidine Assay Preservatives, and Stabilizers Precautions • for in Vitro Diagnostic Use
4 REAgENtS Reagents contain the following substances: Mouse monoclonal antibodies reactive to quinidine (1.35 µg/mL), glucose-6-phosphate (22 mM), nicotinamide adenine dinucleotide (18 mM), quinidine labeled with glucose-6-phosphate dehydrogenase (0.41 U/mL), 0.1% sodium azide, Tris buffer, Quinidine Assay preservatives, and stabilizers Precautions • For in vitro diagnostic use. August 2010 4Q052.2D_B • Contains nonsterile mouse monoclonal antibodies. • Assay components contain sodium azide, which may react with lead and copper plumbing to form highly explosive metal azides. If waste is discarded down the drain, flush it with a large volume of water to prevent azide buildup. • Do not use the kit after the expiration date. • This kit contains streptomycin sulfate. Please dispose of appropriately. • Turbid or yellow reagents may indicate contamination or degradation and must be discarded. Preparation of Reagents Catalog Quantity/ The Emit® 2000 Quinidine Assay reagents are provided ready to use; no preparation is Number Product Description Volume necessary. OSR4Q229 Emit® 2000 Quinidine Assay Storage of Assay Components OSR4Q518 R1 (Antibody/Substrate Reagent 1) 2 x 23 mL • Improper storage of reagents can affect assay performance. OSR4Q548 R2 (Enzyme Reagent 2) 2 x 13 mL • When not in use, store reagents upright at 2–8°C and with screw caps tightly closed. • Unopened reagents are stable until the expiration date printed on the label if stored upright at 4Q109UL Emit® 2000 Quinidine Calibrators* 1 x 5 mL†, 2–8°C. 5 x 2 mL • Do not freeze reagents or expose them to temperatures above 32°C. *Required for calibrating the Emit ® 2000 Quinidine Assay. -

QMS® Quinidine (QUIN)
QMS® Quinidine (QUIN) For In Vitro Diagnostic Use Only 0373936 This Quantitative Microsphere System (QMS) package insert must be read carefully prior to INHALED: If breathing is difficult, remove victim to fresh air and keep at rest in a position use. Package insert instructions must be followed accordingly. Reliability of assay results comfortable for breathing. Specific treatment (see First Aid information on product label cannot be guaranteed if there are any deviations from the instructions in this package insert. and/or Section 4 of the SDS). If skin irritation or rash occurs: Get medical advice/attention. If experiencing respiratory symptoms: Call a POISON CENTER or doctor/physician. Wash Intended Use contaminated clothing before reuse. Dispose of contents/container to location in accordance The QMS® Quinidine assay is intended for the quantitative determination of quinidine in human with local/regional/national/international regulations. serum or plasma on automated clinical chemistry analyzers. CAUTION: This product contains human sourced and/or potentially infectious The results obtained are used in the diagnosis and treatment of quinidine overdose and in components. Components sourced from human blood have been tested and found to be monitoring levels of quinidine to help ensure appropriate therapy. nonreactive for HBsAg, anti-HIV 1/2, and anti-HCV. No known test method can offer complete assurance that products derived from human sources or inactivated microorganisms will Summary and Explanation of the Test not transmit infection. Therefore, it is recommended that all human sourced materials Quinidine is used for the prevention and treatment of ventricular arrhythmias, junctional be considered potentially infectious and handled with appropriate biosafety practices.