Shedding Α TNF- Compensatory Increases in ADAM17 and (ADAM
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

MATRIX METALLOPROTEINASE REQUIREMENTS for NEUROMUSCULAR SYNAPTOGENESIS by Mary Lynn Dear Dissertation Submitted to the Faculty O
MATRIX METALLOPROTEINASE REQUIREMENTS FOR NEUROMUSCULAR SYNAPTOGENESIS By Mary Lynn Dear Dissertation Submitted to the Faculty of the Graduate School of Vanderbilt University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY in Biological Sciences January 31, 2018 Nashville, Tennessee Approved: Todd Graham, Ph.D. Kendal Broadie, Ph.D. Katherine Friedman, Ph.D. Barbara Fingleton, Ph.D. Jared Nordman, Ph.D. Copyright © 2018 by Mary Lynn Dear All Rights Reserved ACKNOWLEDGEMENTS I would like to acknowledge my advisor, Kendal Broadie, and the entire Broadie laboratory, both past and present, for their endless support, engaging conversations, thoughtful suggestions and constant encouragement. I would also like to thank my committee members both past and present for playing such a pivotal role in my graduate career and growth as a scientist. I thank the Department of Biological Sciences for fostering my graduate education. I thank the entire protease community for their continued support, helpful suggestions and collaborative efforts that helped my project move forward. I would like to acknowledge Dr. Andrea Page-McCaw and her entire laboratory for helpful suggestions, being engaged in my studies and providing many tools that were invaluable to my project. I thank my parents, Leisa Justus and Raymond Dear, my brother Jake Dear and my best friend Jenna Kaufman. Words cannot describe how grateful I am for your support and encouragement. Most importantly, I would like to thank my husband, Jeffrey Thomas, for always believing in me, your unwavering support and for helping me endure all the ups and downs that come with graduate school. -

Neprilysin Is Required for Angiotensin-(1-7)
Page 1 of 39 Diabetes NEPRILYSIN IS REQUIRED FOR ANGIOTENSIN-(1-7)’S ABILITY TO ENHANCE INSULIN SECRETION VIA ITS PROTEOLYTIC ACTIVITY TO GENERATE ANGIOTENSIN-(1-2) Gurkirat S. Brara, Breanne M. Barrowa, Matthew Watsonb, Ryan Griesbachc, Edwina Chounga, Andrew Welchc, Bela Ruzsicskad, Daniel P. Raleighb, Sakeneh Zraikaa,c aVeterans Affairs Puget Sound Health Care System, Seattle, WA 98108, United States bDepartment of Chemistry, Stony Brook University, Stony Brook, NY 11794, United States cDivision of Metabolism, Endocrinology and Nutrition, Department of Medicine, University of Washington, Seattle, WA 98195, United States dInstitute for Chemical Biology and Drug Discovery, Stony Brook University, Stony Brook, NY 11794, United States Short Title: Angiotensin-(1-7) and insulin secretion Word count: 3997; Figure count: 8 main (plus 3 Online Suppl.); Table count: 1 Online Suppl. Correspondence to: Sakeneh Zraika, PhD 1660 South Columbian Way (151) Seattle, WA, United States Tel: 206-768-5391 / Fax: 206-764-2164 Email: [email protected] 1 Diabetes Publish Ahead of Print, published online May 30, 2017 Diabetes Page 2 of 39 ABSTRACT Recent work has renewed interest in therapies targeting the renin-angiotensin system (RAS) to improve β-cell function in type 2 diabetes. Studies show that generation of angiotensin-(1-7) by angiotensin converting enzyme 2 (ACE2) and its binding to the Mas receptor (MasR) improves glucose homeostasis, partly by enhancing glucose-stimulated insulin secretion (GSIS). Thus, islet ACE2 upregulation is viewed as a desirable therapeutic goal. Here, we show that although endogenous islet ACE2 expression is sparse, its inhibition abrogates angiotensin-(1-7)-mediated GSIS. However, a more widely expressed islet peptidase, neprilysin, degrades angiotensin-(1-7) into several peptides. -

What Are the Roles of Metalloproteinases in Cartilage and Bone Damage? G Murphy, M H Lee
iv44 Ann Rheum Dis: first published as 10.1136/ard.2005.042465 on 20 October 2005. Downloaded from REPORT What are the roles of metalloproteinases in cartilage and bone damage? G Murphy, M H Lee ............................................................................................................................... Ann Rheum Dis 2005;64:iv44–iv47. doi: 10.1136/ard.2005.042465 enzyme moiety into an upper and a lower subdomain. A A role for metalloproteinases in the pathological destruction common five stranded beta-sheet and two alpha-helices are in diseases such as rheumatoid arthritis and osteoarthritis, always found in the upper subdomain with a further C- and the irreversible nature of the ensuing cartilage and bone terminal helix in the lower subdomain. The catalytic sites of damage, have been the focus of much investigation for the metalloproteinases, especially the MMPs, have been several decades. This has led to the development of broad targeted for the development of low molecular weight spectrum metalloproteinase inhibitors as potential therapeu- synthetic inhibitors with a zinc chelating moiety. Inhibitors tics. More recently it has been appreciated that several able to fully differentiate between individual enzymes have families of zinc dependent proteinases play significant and not been identified thus far, although a reasonable level of varied roles in the biology of the resident cells in these tissues, discrimination is now being achieved in some cases.7 Each orchestrating development, remodelling, and subsequent family does, however, have other unique domains with pathological processes. They also play key roles in the numerous roles, including the determination of physiological activity of inflammatory cells. The task of elucidating the substrate specificity, ECM, or cell surface localisation (fig 1). -

Negatively Regulated by ADAM15 TRIF-Mediated TLR3 and TLR4
TRIF-Mediated TLR3 and TLR4 Signaling Is Negatively Regulated by ADAM15 Suaad Ahmed, Ashwini Maratha, Aisha Qasim Butt, Enda Shevlin and Sinead M. Miggin This information is current as of September 27, 2021. J Immunol 2013; 190:2217-2228; Prepublished online 30 January 2013; doi: 10.4049/jimmunol.1201630 http://www.jimmunol.org/content/190/5/2217 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2013/01/30/jimmunol.120163 Material 0.DC1 References This article cites 57 articles, 20 of which you can access for free at: http://www.jimmunol.org/ http://www.jimmunol.org/content/190/5/2217.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 27, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology TRIF-Mediated TLR3 and TLR4 Signaling Is Negatively Regulated by ADAM15 Suaad Ahmed, Ashwini Maratha, Aisha Qasim Butt, Enda Shevlin, and Sinead M. -

ADAM10 Site-Dependent Biology: Keeping Control of a Pervasive Protease
International Journal of Molecular Sciences Review ADAM10 Site-Dependent Biology: Keeping Control of a Pervasive Protease Francesca Tosetti 1,* , Massimo Alessio 2, Alessandro Poggi 1,† and Maria Raffaella Zocchi 3,† 1 Molecular Oncology and Angiogenesis Unit, IRCCS Ospedale Policlinico S. Martino Largo R. Benzi 10, 16132 Genoa, Italy; [email protected] 2 Proteome Biochemistry, IRCCS San Raffaele Scientific Institute, 20132 Milan, Italy; [email protected] 3 Division of Immunology, Transplants and Infectious Diseases, IRCCS San Raffaele Scientific Institute, 20132 Milan, Italy; [email protected] * Correspondence: [email protected] † These authors contributed equally to this work as last author. Abstract: Enzymes, once considered static molecular machines acting in defined spatial patterns and sites of action, move to different intra- and extracellular locations, changing their function. This topological regulation revealed a close cross-talk between proteases and signaling events involving post-translational modifications, membrane tyrosine kinase receptors and G-protein coupled recep- tors, motor proteins shuttling cargos in intracellular vesicles, and small-molecule messengers. Here, we highlight recent advances in our knowledge of regulation and function of A Disintegrin And Metalloproteinase (ADAM) endopeptidases at specific subcellular sites, or in multimolecular com- plexes, with a special focus on ADAM10, and tumor necrosis factor-α convertase (TACE/ADAM17), since these two enzymes belong to the same family, share selected substrates and bioactivity. We will discuss some examples of ADAM10 activity modulated by changing partners and subcellular compartmentalization, with the underlying hypothesis that restraining protease activity by spatial Citation: Tosetti, F.; Alessio, M.; segregation is a complex and powerful regulatory tool. -

Cell-Autonomous FLT3L Shedding Via ADAM10 Mediates Conventional Dendritic Cell Development in Mouse Spleen
Cell-autonomous FLT3L shedding via ADAM10 mediates conventional dendritic cell development in mouse spleen Kohei Fujitaa,b,1, Svetoslav Chakarovc,1, Tetsuro Kobayashid, Keiko Sakamotod, Benjamin Voisind, Kaibo Duanc, Taneaki Nakagawaa, Keisuke Horiuchie, Masayuki Amagaib, Florent Ginhouxc, and Keisuke Nagaod,2 aDepartment of Dentistry and Oral Surgery, Keio University School of Medicine, Tokyo 160-8582, Japan; bDepartment of Dermatology, Keio University School of Medicine, Tokyo 160-8582, Japan; cSingapore Immunology Network, Agency for Science, Technology and Research, Biopolis, 138648 Singapore; dDermatology Branch, National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health, Bethesda, MD 20892; and eDepartment of Orthopedic Surgery, National Defense Medical College, Tokorozawa 359-8513, Japan Edited by Kenneth M. Murphy, Washington University School of Medicine, St. Louis, MO, and approved June 10, 2019 (received for review November 4, 2018) Conventional dendritic cells (cDCs) derive from bone marrow (BM) intocDC1sorcDC2stakesplaceintheBM(3),andthese precursors that undergo cascades of developmental programs to pre-cDC1s and pre-cDC2s ultimately differentiate into cDC1s terminally differentiate in peripheral tissues. Pre-cDC1s and pre- and cDC2s after migrating to nonlymphoid and lymphoid tissues. + + cDC2s commit in the BM to each differentiate into CD8α /CD103 cDCs are short-lived, and their homeostatic maintenance relies + cDC1s and CD11b cDC2s, respectively. Although both cDCs rely on on constant replenishment from the BM precursors (5). The cy- the cytokine FLT3L during development, mechanisms that ensure tokine Fms-related tyrosine kinase 3 ligand (FLT3L) (12), by cDC accessibility to FLT3L have yet to be elucidated. Here, we gen- signaling through its receptor FLT3 expressed on DC precursors, erated mice that lacked a disintegrin and metalloproteinase (ADAM) is essential during the development of DCs (7, 13). -

Expression of the Disintegrin Metalloprotease, ADAM-10, In
314 Vol. 10, 314–323, January 1, 2004 Clinical Cancer Research Expression of the Disintegrin Metalloprotease, ADAM-10, in Prostate Cancer and Its Regulation by Dihydrotestosterone, Insulin-Like Growth Factor I, and Epidermal Growth Factor in the Prostate Cancer Cell Model LNCaP Daniel R. McCulloch,1 Pascal Akl,1 Conclusions: This study describes for the first time Hemamali Samaratunga,2 Adrian C. Herington,1 the expression, regulation, and cellular localization of and Dimitri M. Odorico1 ADAM-10 protein in PCa. The regulation and membrane 1 localization of ADAM-10 support our hypothesis that Hormone-Dependent Cancer Program, School of Life Sciences, ADAM-10 has a role in extracellular matrix maintenance Queensland University of Technology, Brisbane, Queensland, Australia, and 2Sullivan Nicolaides Pathology, Brisbane, Queensland, and cell invasion, although the potential role of nuclear Australia ADAM-10 is not yet known. INTRODUCTION ABSTRACT The disintegrin metalloproteases ADAMs, like the matrix Purpose: The disintegrin metalloprotease ADAM-10 is metalloproteinases (MMPs), are members of the metzincin a multidomain metalloprotease that is potentially significant (zinc-dependent metalloprotease) superfamily. To date, more in tumor progression due to its extracellular matrix-degrad- than 30 ADAMs have been characterized (1), some of which are ing properties. Previously, ADAM-10 mRNA was detected involved in diverse biological functions such as fertilization, in prostate cancer (PCa) cell lines; however, the presence of neurogenesis (2, 3), and the ectodomain shedding of growth ADAM-10 protein and its cellular localization, regulation, factors such as amyloid precursor protein and tumor necrosis and role have yet to be described. We hypothesized that factor ␣ (4, 5). -

ADAM17 Targets MMP-2 and MMP-9 Via EGFR-MEK-ERK Pathway Activation to Promote Prostate Cancer Cell Invasion
1714 INTERNATIONAL JOURNAL OF ONCOLOGY 40: 1714-1724, 2012 ADAM17 targets MMP-2 and MMP-9 via EGFR-MEK-ERK pathway activation to promote prostate cancer cell invasion LI-JIE XIAO1,2*, PING LIN1*, FENG LIN1*, XIN LIU1, WEI QIN1, HAI-FENG ZOU1, LIANG GUO1, WEI LIU1, SHU-JUAN WANG1 and XIAO-GUANG YU1 1Department of Biochemistry and Molecular Biology, College of Basic Medical Science, Harbin Medical University, 194 Xuefu Road, Harbin 150081, Heilongjiang; 2College of Life Science and Technology, Heilongjiang Bayi Agricultural University, 2 Xinyang Road, Daqing 163319, Heilongjiang, P.R. China Received September 8, 2011; Accepted November 18, 2011 DOI: 10.3892/ijo.2011.1320 Abstract. ADAM17, also known as tumor necrosis factor-α released and down-regulation of MMP-2, MMP-9. However, converting enzyme (TACE), is involved in proteolytic ectodo- these effects could be reversed by simultaneous addition of main shedding of cell surface molecules and cytokines. TGF-α. These data demonstrated that ADAM17 contributes to Although aberrant expression of ADAM17 has been shown androgen-independent prostate cancer cell invasion by shed- in various malignancies, the function of ADAM17 in prostate ding of EGFR ligand TGF-α, which subsequently activates the cancer has not been clarified. In the present study, we sought EGFR-MEK-ERK signaling pathway, leading finally to over- to elucidate whether ADAM17 contributes to prostate cancer expression of MMP-2 and MMP-9. This study suggests that the cell invasion, as well as the mechanism involved in the process. ADAM17 expression level may be a new predictive biomarker The expression pattern of ADAM17 was investigated in human of invasion and metastasis of prostate cancer, and ADAM17 prostate cancer cells. -
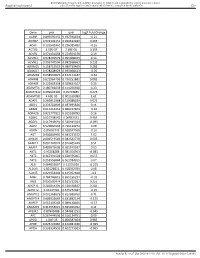
Gene Pval Qval Log2 Fold Change AAMP 0.895690332 0.952598834
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut Gene pval qval Log2 Fold Change AAMP 0.895690332 0.952598834 -0.21 ABI3BP 0.002302151 0.020612283 0.465 ACHE 0.103542461 0.296385483 -0.16 ACTG2 2.99E-07 7.68E-05 3.195 ACVR1 0.071431098 0.224504378 0.19 ACVR1C 0.978209579 0.995008423 0.14 ACVRL1 0.006747504 0.042938663 0.235 ADAM15 0.158715519 0.380719469 0.285 ADAM17 0.978208929 0.995008423 -0.05 ADAM28 0.038932876 0.152174187 -0.62 ADAM8 0.622964796 0.790251882 0.085 ADAM9 0.122003358 0.329623107 0.25 ADAMTS1 0.180766659 0.414256926 0.23 ADAMTS12 0.009902195 0.05703885 0.425 ADAMTS8 4.60E-05 0.001169089 1.61 ADAP1 0.269811968 0.519388039 0.075 ADD1 0.233702809 0.487695826 0.11 ADM2 0.012213453 0.066227879 -0.36 ADRA2B 0.822777921 0.915518785 0.16 AEBP1 0.010738542 0.06035531 0.465 AGGF1 0.117946691 0.320915024 -0.095 AGR2 0.529860903 0.736120272 0.08 AGRN 0.85693743 0.928047568 -0.16 AGT 0.006849995 0.043233572 1.02 AHNAK 0.006519543 0.042542779 0.605 AKAP12 0.001747074 0.016405449 0.51 AKAP2 0.409929603 0.665919397 0.05 AKT1 0.95208288 0.985354963 -0.085 AKT2 0.367391504 0.620376005 0.055 AKT3 0.253556844 0.501934205 0.07 ALB 0.064833867 0.21195036 -0.315 ALDOA 0.83128831 0.918352939 0.08 ALOX5 0.029954404 0.125352668 -0.3 AMH 0.784746815 0.895196237 -0.03 ANG 0.050500474 0.181732067 0.255 ANGPT1 0.281853305 0.538528647 0.285 ANGPT2 0.43147281 0.675272487 -0.15 ANGPTL2 0.001368876 0.013688762 0.71 ANGPTL4 0.686032669 0.831882134 -0.175 ANPEP 0.019103243 0.089148466 -0.57 ANXA2P2 0.412553021 0.665966092 0.11 AP1M2 0.87843088 0.944681253 -0.045 APC 0.267444505 0.516134751 0.09 APOD 1.04E-05 0.000587404 0.985 APOE 0.023722987 0.104981036 -0.395 APOH 0.336334555 0.602273505 -0.065 Sundar R, et al. -

Gene Standard Deviation MTOR 0.12553731 PRPF38A
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut Gene Standard Deviation MTOR 0.12553731 PRPF38A 0.141472605 EIF2B4 0.154700091 DDX50 0.156333027 SMC3 0.161420017 NFAT5 0.166316903 MAP2K1 0.166585267 KDM1A 0.16904912 RPS6KB1 0.170330192 FCF1 0.170391706 MAP3K7 0.170660513 EIF4E2 0.171572093 TCEB1 0.175363093 CNOT10 0.178975095 SMAD1 0.179164705 NAA15 0.179904998 SETD2 0.180182498 HDAC3 0.183971158 AMMECR1L 0.184195031 CHD4 0.186678211 SF3A3 0.186697697 CNOT4 0.189434633 MTMR14 0.189734199 SMAD4 0.192451524 TLK2 0.192702667 DLG1 0.19336621 COG7 0.193422331 SP1 0.194364189 PPP3R1 0.196430217 ERBB2IP 0.201473001 RAF1 0.206887192 CUL1 0.207514271 VEZF1 0.207579584 SMAD3 0.208159809 TFDP1 0.208834504 VAV2 0.210269344 ADAM17 0.210687138 SMURF2 0.211437666 MRPS5 0.212428684 TMUB2 0.212560675 SRPK2 0.216217428 MAP2K4 0.216345366 VHL 0.219735582 SMURF1 0.221242495 PLCG1 0.221688351 EP300 0.221792349 Sundar R, et al. Gut 2020;0:1–10. doi: 10.1136/gutjnl-2020-320805 BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut MGAT5 0.222050228 CDC42 0.2230598 DICER1 0.225358787 RBX1 0.228272533 ZFYVE16 0.22831803 PTEN 0.228595789 PDCD10 0.228799406 NF2 0.23091035 TP53 0.232683696 RB1 0.232729172 TCF20 0.2346075 PPP2CB 0.235117302 AGK 0.235416298 -

Matrix Metalloproteinases in Angiogenesis: a Moving Target for Therapeutic Intervention
Matrix metalloproteinases in angiogenesis: a moving target for therapeutic intervention William G. Stetler-Stevenson J Clin Invest. 1999;103(9):1237-1241. https://doi.org/10.1172/JCI6870. Perspective Angiogenesis is the process in which new vessels emerge from existing endothelial lined vessels. This is distinct from the process of vasculogenesis in that the endothelial cells arise by proliferation from existing vessels rather than differentiating from stem cells. Angiogenesis is an invasive process that requires proteolysis of the extracellular matrix and, proliferation and migration of endothelial cells, as well as synthesis of new matrix components. During embryonic development, both vasculogenesis and angiogenesis contribute to formation of the circulatory system. In the adult, with the single exception of the reproductive cycle in women, angiogenesis is initiated only in response to a pathologic condition, such as inflammation or hypoxia. The angiogenic response is critical for progression of wound healing and rheumatoid arthritis. Angiogenesis is also a prerequisite for tumor growth and metastasis formation. Therefore, understanding the cellular events involved in angiogenesis and the molecular regulation of these events has enormous clinical implications. This understanding is providing novel therapeutic targets for the treatment of a variety of diseases, including cancer. Whatever the pathologic condition, an initiating stimulus results in the formation of a migrating solid column of endothelial cells called the vascular sprout. The advancing front of this endothelial cell column presumably focuses proteolytic activity to create a defect in the extracellular matrix, through which the advancing and proliferating column of endothelial […] Find the latest version: https://jci.me/6870/pdf Matrix metalloproteinases in angiogenesis: Perspective a moving target for therapeutic intervention SERIES Topics in angiogenesis David A. -

CD Markers Are Routinely Used for the Immunophenotyping of Cells
ptglab.com 1 CD MARKER ANTIBODIES www.ptglab.com Introduction The cluster of differentiation (abbreviated as CD) is a protocol used for the identification and investigation of cell surface molecules. So-called CD markers are routinely used for the immunophenotyping of cells. Despite this use, they are not limited to roles in the immune system and perform a variety of roles in cell differentiation, adhesion, migration, blood clotting, gamete fertilization, amino acid transport and apoptosis, among many others. As such, Proteintech’s mini catalog featuring its antibodies targeting CD markers is applicable to a wide range of research disciplines. PRODUCT FOCUS PECAM1 Platelet endothelial cell adhesion of blood vessels – making up a large portion molecule-1 (PECAM1), also known as cluster of its intracellular junctions. PECAM-1 is also CD Number of differentiation 31 (CD31), is a member of present on the surface of hematopoietic the immunoglobulin gene superfamily of cell cells and immune cells including platelets, CD31 adhesion molecules. It is highly expressed monocytes, neutrophils, natural killer cells, on the surface of the endothelium – the thin megakaryocytes and some types of T-cell. Catalog Number layer of endothelial cells lining the interior 11256-1-AP Type Rabbit Polyclonal Applications ELISA, FC, IF, IHC, IP, WB 16 Publications Immunohistochemical of paraffin-embedded Figure 1: Immunofluorescence staining human hepatocirrhosis using PECAM1, CD31 of PECAM1 (11256-1-AP), Alexa 488 goat antibody (11265-1-AP) at a dilution of 1:50 anti-rabbit (green), and smooth muscle KD/KO Validated (40x objective). alpha-actin (red), courtesy of Nicola Smart. PECAM1: Customer Testimonial Nicola Smart, a cardiovascular researcher “As you can see [the immunostaining] is and a group leader at the University of extremely clean and specific [and] displays Oxford, has said of the PECAM1 antibody strong intercellular junction expression, (11265-1-AP) that it “worked beautifully as expected for a cell adhesion molecule.” on every occasion I’ve tried it.” Proteintech thanks Dr.