Overview of Comments on EMA/641479/2014 Draft Proposal for an Addendum, on Transparency, to the “Functional Specifications
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Addendum/Erratum for Elliptic Curves 2006
Addendum/Erratum for Elliptic Curves 2006 J.S. Milne Last revised September 7, 2016. In the blurb and introduction, I should have noted that the group is commutative. p28. The third cubic curve should be `.R;Q/ `.P;Q R/ `.PQ;O/ 0 C D (Dmitriy Zanin). p36. In the definition of kŒC p, the condition on h should be h p (Jochen Gerhard). … p39. In the definition of a regular map between projective plane curves, am should read a2 (Rankeya Datta). p100, 3.23b. The sign is wrong: it should read 4d c2 0. As PENG Bo pointed out to me, I forgot to include the proof. Here it is. Let X 2 c X d det.X n˛ T E/: C 0 C 0 D j ` By linear algebra, we see that c nc and d n2d. On substituting m for X in the equality, we 0 D 0 D find that m2 cmn n2d det.m n˛ T E/: C C D j ` According to Proposition 3.22, the right hand side equals the degree of mid n˛. Therefore m2 cmn n2d 0 C C for all m;n Z, i.e., 2 r2 cr d 0 C C 2 c 2 c c2 for all r Q. The minimum value of r cr d; r R, is . / c. / d d, and so 2 C C 2 2 C 2 C D 4 C 4d c2 (happily, this is how I used it on p150 in the proof of the congruence Riemann hypothesis). p107, line 2 (exact sequence of cohomology groups): a bracket “/” is missing: H 1.G;.kal // instead of H 1.G;.kal / (Michael Mueller). -
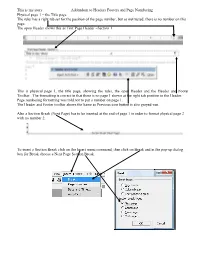
This Is My Story Addendum to Headers Footers and Page Numbering Physical Page 1 – the Title Page the Ruler Has a Right
This is my story Addendum to Headers Footers and Page Numbering Physical page 1 – the Title page The ruler has a right tab set for the position of the page number, but as instructed, there is no number on this page. The open Header shows this as First Page Header –Section 1. This is physical page 1, the title page, showing the ruler, the open Header and the Header and Footer Toolbar. The formatting is correct in that there is no page 1 shown at the right tab position in the Header. Page numbering formatting was told not to put a number on page 1. The Header and Footer toolbar shows the Same as Previous icon button is also greyed-out. Also a Section Break (Next Page) has to be inserted at the end of page 1 in order to format physical page 2 with no number 2. To insert a Section Break click on the Insert menu command, then click on Break and in the pop-up dialog box for Break choose a Next Page Section Break. This is my story Addendum to Headers Footers and Page Numbering This is physical page 2 – the Acknowledgement/Dedication page. This is now First Page Header –Section 2-. This is physical page 2, the Dedication page, showing the ruler, the open Header and the Header and Footer Toolbar. Again the formatting is correct in that there is no page 2 shown at right-tab position in the Header. In the Header and Footer toolbar the Same as Previous icon button is depressed. -

Postscript Printer Description (PPD)
PostScript Printer Description File Format POSTSCRIPTR Specification Software From Adboe Adobe Developer Support Version 4.3 9 February 1996 Adobe Systems Incorporated Adobe Developer Technologies 345 Park Avenue San Jose, CA 95110 http://partners.adobe.com/ PN LPS5003 Copyright 1987-1996 by Adobe Systems Incorporated. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording, or otherwise, without the prior written consent of the publisher. Any software referred to herein is furnished under license and may only be used or copied in accordance with the terms of such license. PostScript is a registered trademark of Adobe Systems Incorporated. All instances of the name PostScript in the text are references to the PostScript language as defined by Adobe Systems Incorporated unless otherwise stated. The name PostScript also is used as a product trademark for Adobe Systems’ implementation of the PostScript language interpreter. Any references to a “PostScript printer,” a “PostScript file,” or a “PostScript driver” refer to printers, files, and driver programs (respectively) which are written in or support the PostScript language. The sentences in this book that use “PostScript language” as an adjective phrase are so constructed to reinforce that the name refers to the standard language definition as set forth by Adobe Systems Incorporated. PostScript, the PostScript logo, Display PostScript, Adobe, and the Adobe logo are trademarks of Adobe Systems Incorporated which may be registered in certain jurisdictions. Apple, AppleTalk, LaserWriter, and Macintosh are registered trademarks of Apple Computer, Inc. -

Greco RC-II 2006 9E Final Addendum Belgium PUBLIC
DIRECTORATE GENERAL OF HUMAN RIGHTS AND LEGAL AFFAIRS DIRECTORATE OF MONITORING Strasbourg, 15 May 2009 Public Greco RC-II (2006) 9E Addendum Second Evaluation Round Addendum to the Compliance Report on Belgium Adopted by GRECO at its 42 nd Plenary Meeting (Strasbourg, 11-15 May 2009) Secrétariat du GRECO GRECO Secretariat www.coe.int/greco Conseil de l’Europe Council of Europe F-67075 Strasbourg Cedex +33 3 88 41 20 00 Fax +33 3 88 41 39 55 I. INTRODUCTION 1. GRECO adopted the Second Round Evaluation Report on Belgium at its 21 st Plenary Meeting (2 December 2004). The report (Greco Eval II Rep (2004) 1E) was made public by GRECO on 24 January 2005, following authorisation from the Belgian authorities. 2. Belgium submitted the situation report required under GRECO's compliance procedure on 8 June 2006. On the basis of this report, and following a discussion in plenary session, GRECO adopted the Second Round Compliance Report (RC report) on Belgium at its 33 rd Plenary Meeting (1 June 2007). It was made public on 25 June 2007. The compliance report (Greco RC-II (2006) 9E) concluded that recommendations ii and vii had been implemented satisfactorily. Recommendation i had been dealt with in a satisfactory manner. Recommendations iv, v, vi and ix had been partly implemented and recommendations iii and viii had not been implemented. GRECO requested additional information on their implementation. This information was submitted to it on 2 December 2008, and on 18 March 2009 in a supplementary report. 3. Pursuant to Rule 31, paragraph 9.1 of GRECO's Rules of Procedure, the purpose of this addendum to the second round compliance report is to assess the implementation of recommendations iii, iv, v, vi, viii and ix in the light of the additional information referred to in paragraph 2. -

Digital Appellate Record Standards for the Supreme Court of Virginia and Court of Appeals of Virginia
Digital Appellate Record Standards for the Supreme Court of Virginia and Court of Appeals of Virginia A. The Digital Appellate Record ("DAR"). 1. The Digital Appellate Record is a collection of Portable Document Format (PDF) files and documents. a. A PDF file is a document that may include multiple pages. b. The DAR may contain multiple PDFs, e.g. Manuscripts, Transcripts and Exhibits. 2. The DAR will be separated into the following PDF files with an individual Table of Contents and a number designation preceding each PDF file name. a. 00 Master Table of Contents b. 01 Manuscripts c. 02 Transcripts d. 03 Exhibits 3. Depositions will be included in the DAR either 1) in the manuscript or 2) as an exhibit, if filed as such. 4. All DARs will be submitted via a Digital Record System (DRS). Any file larger than 150MB will be automatically rejected. 5. Tribunals should retain a copy of each DAR submitted to an Appellate Court. Tribunals are also required to retain the original documents filed in the case until (1) the expiration of 70 days from the date of the final order, if no notice of appeal has been filed, or (2) all appeals to the Court of Appeals of Virginia and the Supreme Court of Virginia are final, if a notice of appeal has been filed. 6. Scanning specifications – the scanned page should be a complete and accurate representation of the original. (Adobe Acrobat 8 Standard or above is suggested): a. Color Mode i. Most documents will be scanned in black and white. -

BOSTON BOOK FAIR ADDENDUM a Short List E-Catalogue
BOSTON BOOK FAIR ADDENDUM a short list e-catalogue Donald A. Heald | Rare Books Donald A. Heald | Rare Books BOSTON BOOK FAIR ADDENDUM a short list e-catalogue Just click the title of each item in this illustrated short list and follow the link to a full description and images on our website Questions? Contact us or email [email protected] DDoonnaalldd AA.. HHeeaalldd || RRaarree BBooookkss CLICK THE TITLE JOHN JAMES AUDUBON Autograph manuscript, from Audubon's Ornithological Biography, recounting his meeting with Thomas Bewick [Great Britain: 1830-1835]. 3pp. on two foolscap sheets. Expert repairs to separations at folds. Housed in a red morocco backed box. Autograph manuscript by Audubon, on meeting Thomas Bewick. (#34588) $ 15,000 DDoonnaalldd AA.. HHeeaalldd || RRaarree BBooookkss CLICK THE TITLE BROADSIDE By His Excellency Francis Bernard, Esq; Captain General in Chief in and over His Majesty's Province of the Massachusetts-Bay, in New-England, and Vice-Admiral of the same. A Proclamation for a general Fast Boston: Richard Draper, 1767. Letterpress broadside, 15 1/2 x 13 inches. Woodcut royal arms, signed in print by Bernard. Minor repairs at folds on verso. Early American broadside. (#34640) $ 1,500 DDoonnaalldd AA.. HHeeaalldd || RRaarree BBooookkss CLICK THE WILLIAM TITLE COBBETT The American Gardener; or a treatise on the situation, soil fencing and laying-out of gardens; on the making and managing of hot- beds and green-houses; and on the propagation and cultivation of the several sorts of vegetables, herbs, fruits and flowers London: C. Clement, 1821. 12mo (7 3/8 x 4 1/4 inches). -

Bay Leaf Used & Rare Books
Bay Leaf Used & Rare Books Bookplate Literature & Bookplates Catalogue Four December 2012 Bay Leaf Used & Rare Books G.L. Konrád, Bookseller 49 E. Lake, PO Box 105, Sand Lake, MI 49343 (616) 636-8500 [email protected] www.bayleafbooks.com Thank you for taking time to explore our catalogue; please feel free to call or email with any questions. All items subject to prior sale; please call or email to reserve. Unless otherwise stated, signed volumes do not have inscriptions. Photographs can be emailed upon request. Terms: All items are packed and posted with care. Domestic shipping via Media Mail is $3.50 for the first item, and $1.00 for each additional item (adjustments are made for small booklets, pamphlets, bookplates, etc.). Priority rates are available. Foreign shipping is billed at cost. Payment is accepted via MasterCard, Visa, Discover, PayPal, money orders or checks (U.S. funds drawn on a U.S. bank). Trade discounts are available and institutional purchase orders are welcome. Michigan residents must add 6% sales tax. Approved, prompt returns accepted. On the cover: Bookplate designed for Gabe Konrád, proprietor of Bay Leaf Books, by Daniel Mitsui (www.danielmitsui.com) and printed on Rives Light paper by Chad Pastotnik of Deep Wood Press (www.deepwoodpress.com). We are members of the Antiquarian Booksellers Association of America, the International League of Antiquarian Booksellers, and the Independent Online Booksellers Association and adhere to those organizations’ standards of professionalism and ethics. 4001. Allen, Charles Dexter. A Classified List of Early American Book-Plates with a Brief Description of the Principal Styles and a Note as to the Promi- nent Engravers. -

CITY of SAMMAMISH SURFACE WATER DESIGN MANUAL ADDENDUM PREFACE – How to Use This Document
ATTACHMENT “B” CITY OF SAMMAMISH SURFACE WATER DESIGN MANUAL ADDENDUM PREFACE – How to Use this Document General Introduction This document was prepared for the City of Sammamish to meet the requirements of the NPDES Phase 2 permit to develop, implement, and enforce a program to reduce pollutants in stormwater runoff from new development, redevelopment and construction site activities. Per the NPDES permit, this program applies to all sites that disturb a land area 1 acre or greater (the NPDES regulatory threshold), including projects less than one acre that are part of a larger common plan of the development or sale. The program applies to private and public development, including roads. The City of Sammamish has adopted the 2009 King County Surface Water Design Manual (2009 KCSWDM) in order to be in compliance with its Phase II Municipal Stormwater permit. This Addendum to the 2009 KCSWDM defines how the requirements of the KCSWDM are to be implemented within the City of Sammamish. The Addendum specifies all changes, additions, and deletions to the 2009 KCSWDM to make it appropriate for use within the City of Sammamish. The 2009 KCSWDM along with this Addendum define the drainage requirements for development and redevelopment projects within the City. Ecology has allowed local jurisdictions to follow previous stormwater requirements for projects disturbing less than one acre. The City has elected to continue their previous practice of applying the requirements of the 1998 King County Surface Water Design Manual (1998 KCSWDM) to those projects disturbing less than the required threshold (1 acre). Which Manual should be used If your project will disturb less than acre and is not part of a larger common plan or sale, then use the 1998 KCSWDM in conjunction with the applicable sections of this addendum. -

Accounts Receivable
RESOURCE AND PATIENT MANAGEMENT SYSTEM Accounts Receivable (BAR) Addendum to User Manual Version 1.8 Patch 29 July 2020 Office of Information Technology Division of Information Technology Accounts Receivable (BAR) Version 1.8 Patch 29 Table of Contents 1.0 Introduction ......................................................................................................... 1 1.1 Summary of Changes .............................................................................. 1 1.1.1 Patch 29 ................................................................................................ 1 2.0 Patch 29 ............................................................................................................... 4 2.1 Electronic Remittance Advice (835 ERA) ................................................ 4 2.1.1 Load New Import ................................................................................... 4 2.1.2 Review and Match ERA to Collection Batch .......................................... 5 2.1.3 A/R Bill Matching ................................................................................... 6 2.1.4 Review Postable Claims ........................................................................ 7 2.1.5 Report ERA Claims ............................................................................... 8 2.2 ERA Processing for Tribal/638/Urban Facilities ....................................... 9 2.3 Reports .................................................................................................. 12 2.3.1 Top Payer -

Newsletter No. 30
THE NEW AUSTRALIAN BOOKPLATE SOCIETY collectors, bibliophiles, artists and others dedicated to promoting bookplates Newsletter No. 30, September 2013 Editor/President The Society’s sixth Annual General Meeting Dr Mark Ferson 4 Sofala Ave Riverview NSW 2066 This year the Society’s Annual General Meeting engraving. John discussed a number of his 02 9428 2863 was held on the evening of 29 May in the older armorial and pictorial designs as well as [email protected] Conference Room of the iconic Dymocks contemporary ones, including bookplates for Secretary Building in Sydney’s central business district, in members of the Waterhouse horse-racing family Bronwyn Vost honour of our guest speaker, John W Thompson, and a design for Sydney polymath and ABC 59 Gladstone St, who occupies a suite in the building – and radio host Adam Spencer. After John fielded Enmore NSW 2042 whose good offices had led to the Society being many questions from spell-bound members, it [email protected] able to use the venue. John is Sydney’s only was reluctantly decided that we needed to move hand engraver and specialises in the engraving onto the business of the meeting. Our speaker of jewellery and presentation items but also was thanked heartily for his fascinating talk and strays into other uses including the decoration presented with a small gift. of antique firearms and, of relevance to our The President gave his report for 2012, in Society, the execution of copper engravings which he highlighted the ongoing work on for the production of bookplates. John spoke attempting to keep the Soicety’s website up to prior to the conduct of official Society business, date, the continuing success of the Newsletter relating how he first became involved in art and the publication of the Directory of members ‘back home’ in England and undertook an 2011. -

Epilogue: Ramsey’S Ubiquitous Pragmatism
10 Epilogue: Ramsey’s Ubiquitous Pragmatism HUW PRICE A FEW WEEKS BEFORE I ARRIVED in Cambridge in 2011, as Simon Blackburn’s suc cessor in the newly named Bertrand Russell Professorship, there was an oppor tunity to apply for conference funding from a new University scheme. With Fraser MacBride, I came up with a proposal that seemed not only an excellent fit with our own interests, but astonishingly inclusive within recent Cambridge philoso phy more generally. From my side, it connected my work not only to Blackburn (that link was obvious) and to several of his and my apparently disparate prede cessors in the chair, such as Mellor, Anscombe, von Wright, and Wittgenstein, but also to many other distinguished Cambridge philosophers of the past century or so—Frank Ramsey, Bernard Williams, and Edward Craig, for example. (From MacBride’s side, it connected to his interest in neglected figures in Cambridge philosophy in the interwar period.) What was this appealingly broad church? It was the view that for some in teresting topics, the path to philosophical illumination lies not, as others have thought, in an inquiry into the nature of the (apparent) subject matter, but in asking about the distinctive role of the concepts in question—how we come to have such concepts, what roles they play in our lives, and so on. This view is very famil iar in Blackburn’s work on topics such as morality and modality, for example— Blackburn now calls it expressivism, and traces it in both these cases to Hume. But it also turns up, in places, in the work of a very wide range of other Cambridge philosophers: at least arguably, for example, in that of Mellor on tense, Anscombe on the first person, Craig on knowledge, von Wright on causation, Williams on truth, as well as Wittgenstein and Ramsey, famously, on various matters. -

AUSTRALIAN BOOKPLATE SOCIETY Collectors, Bibliophiles, Artists and Others Dedicated to Promoting Bookplates Newsletter No
THE NEW AUSTRALIAN BOOKPLATE SOCIETY collectors, bibliophiles, artists and others dedicated to promoting bookplates Newsletter No. 51, December 2018 Editor/President The 37th FISAE Congress in Prague, 2018 Dr Mark Ferson 4 Sofala Ave By Andrew Peake, Adelaide Riverview NSW 2066 0401 141 890 The International Ex Libris Congress was held A very full social program had also been [email protected] in Prague, Czech Republic, between 28 August organised: a tram tour of the city, a tour of the Secretary and 1 September, at the Pyramida Hotel, Prague Castle and Strahov Monastery, smaller Bronwyn Vost walking distance from the Prague Castle. By tours to parts of Prague, a trip to Karlstein [email protected] the end of April there were 215 registrants, Castle and a post-congress tour to Marienbad. Designer but the final number grew to 544 participants, In addition to the tourist-type opportunities, Mary Keep one of the largest congresses ever. However, there were a number of exhibitions organised [email protected] my wife and I were the only participants from off-site, the main one being the exhibition Australia, and there was one attendee from of competition entries for which a book was New Zealand and two from Argentina for the published. The others were a retrospective of the southern hemisphere. As to be expected, the ex libris of Vladimir Schanek (1951-2018) at bulk of the participants were from Europe, Marienbad, and the ‘History of Czech ex libris although a significant number came from until 1945’ at the National Memorial, where China, Hong Kong and Turkey.