Linkage/Association Study of a Locus Modulating Total Serum Ige On
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Seq2pathway Vignette
seq2pathway Vignette Bin Wang, Xinan Holly Yang, Arjun Kinstlick May 19, 2021 Contents 1 Abstract 1 2 Package Installation 2 3 runseq2pathway 2 4 Two main functions 3 4.1 seq2gene . .3 4.1.1 seq2gene flowchart . .3 4.1.2 runseq2gene inputs/parameters . .5 4.1.3 runseq2gene outputs . .8 4.2 gene2pathway . 10 4.2.1 gene2pathway flowchart . 11 4.2.2 gene2pathway test inputs/parameters . 11 4.2.3 gene2pathway test outputs . 12 5 Examples 13 5.1 ChIP-seq data analysis . 13 5.1.1 Map ChIP-seq enriched peaks to genes using runseq2gene .................... 13 5.1.2 Discover enriched GO terms using gene2pathway_test with gene scores . 15 5.1.3 Discover enriched GO terms using Fisher's Exact test without gene scores . 17 5.1.4 Add description for genes . 20 5.2 RNA-seq data analysis . 20 6 R environment session 23 1 Abstract Seq2pathway is a novel computational tool to analyze functional gene-sets (including signaling pathways) using variable next-generation sequencing data[1]. Integral to this tool are the \seq2gene" and \gene2pathway" components in series that infer a quantitative pathway-level profile for each sample. The seq2gene function assigns phenotype-associated significance of genomic regions to gene-level scores, where the significance could be p-values of SNPs or point mutations, protein-binding affinity, or transcriptional expression level. The seq2gene function has the feasibility to assign non-exon regions to a range of neighboring genes besides the nearest one, thus facilitating the study of functional non-coding elements[2]. Then the gene2pathway summarizes gene-level measurements to pathway-level scores, comparing the quantity of significance for gene members within a pathway with those outside a pathway. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

The Enigmatic Charcot-Leyden Crystal Protein
C al & ellu ic la n r li Im C m f u Journal of o n l o a l n o r Clarke et al., J Clin Cell Immunol 2015, 6:2 g u y o J DOI: 10.4172/2155-9899.1000323 ISSN: 2155-9899 Clinical & Cellular Immunology Review Article Open Access The Enigmatic Charcot-Leyden Crystal Protein (Galectin-10): Speculative Role(s) in the Eosinophil Biology and Function Christine A Clarke1,3, Clarence M Lee2 and Paulette M Furbert-Harris1,3,4* 1Department of Microbiology, Howard University College of Medicine, Washington, D.C., USA 2Department of Biology, Howard University College of Arts and Sciences, Washington, D.C., USA 3Howard University Cancer Center, Howard University College of Medicine, Washington, D.C., USA 4National Human Genome Center, Howard University College of Medicine, Washington, D.C., USA *Corresponding author: Furbert-Harris PM, Department of Microbiology, Howard University Cancer Center, 2041 Georgia Avenue NW, Room #530, 20060, Washington, D.C., USA, Tel: 202-806-7722, Fax: 202-667-1686; Email: [email protected] Received date: Febuary 22, 2015, Accepted date: April 25, 2015, Published date: April 29, 2015 Copyright: © 2015 Clarke CA, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract Eosinophilic inflammation in peripheral tissues is typically marked by the deposition of a prominent eosinophil protein, Galectin-10, better known as Charcot-Leyden crystal protein (CLC). Unlike the eosinophil’s four distinct toxic cationic proteins and enzymes [major basic protein (MBP), eosinophil-derived neurotoxin (EDN), eosinophil cationic protein (ECP), and eosinophil peroxidase (EPO)], there is a paucity of information on the precise role of the crystal protein in the biology of the eosinophil. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

Human Lectins, Their Carbohydrate Affinities and Where to Find Them
biomolecules Review Human Lectins, Their Carbohydrate Affinities and Where to Review HumanFind Them Lectins, Their Carbohydrate Affinities and Where to FindCláudia ThemD. Raposo 1,*, André B. Canelas 2 and M. Teresa Barros 1 1, 2 1 Cláudia D. Raposo * , Andr1 é LAQVB. Canelas‐Requimte,and Department M. Teresa of Chemistry, Barros NOVA School of Science and Technology, Universidade NOVA de Lisboa, 2829‐516 Caparica, Portugal; [email protected] 12 GlanbiaLAQV-Requimte,‐AgriChemWhey, Department Lisheen of Chemistry, Mine, Killoran, NOVA Moyne, School E41 of ScienceR622 Co. and Tipperary, Technology, Ireland; canelas‐ [email protected] NOVA de Lisboa, 2829-516 Caparica, Portugal; [email protected] 2* Correspondence:Glanbia-AgriChemWhey, [email protected]; Lisheen Mine, Tel.: Killoran, +351‐212948550 Moyne, E41 R622 Tipperary, Ireland; [email protected] * Correspondence: [email protected]; Tel.: +351-212948550 Abstract: Lectins are a class of proteins responsible for several biological roles such as cell‐cell in‐ Abstract:teractions,Lectins signaling are pathways, a class of and proteins several responsible innate immune for several responses biological against roles pathogens. such as Since cell-cell lec‐ interactions,tins are able signalingto bind to pathways, carbohydrates, and several they can innate be a immuneviable target responses for targeted against drug pathogens. delivery Since sys‐ lectinstems. In are fact, able several to bind lectins to carbohydrates, were approved they by canFood be and a viable Drug targetAdministration for targeted for drugthat purpose. delivery systems.Information In fact, about several specific lectins carbohydrate were approved recognition by Food by andlectin Drug receptors Administration was gathered for that herein, purpose. plus Informationthe specific organs about specific where those carbohydrate lectins can recognition be found by within lectin the receptors human was body. -
Next-MP50 Status Report
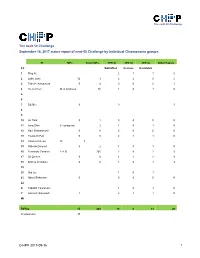
The neXt-50 Challenge The neXt-50 Challenge September 16, 2017 status report of next-50 Challenge by individual Chromosome groups. PI MPs Silver MPs JPR SI JPR SI JPR SI Other Papers Ch Submitted In press In revision 1 Ping Xu 2 1 1 0 2 Lydie Lane 12 3 2 2 0 2 3 Takeshi Kawamura 0 4 0 0 0 1 4 Yu-Ju Chen 26 in progress 86 1 0 1 0 5 6 7 Ed Nice 0 0 1 8 9 10 Jin Park 0 1 0 0 0 0 11 Jong Shin 2 in progress 2 1 0 1 0 12 Ravi Sirdeshmukh 0 0 0 0 0 0 13 Young-Ki Paik 0 5 2 1 1 0 14 CharLes Pineau 12 3 15 GiLberto Domont 0 2 1 0 1 0 16 Fernando CorraLes 2 (+ 9) 165 2 0 2 5 17 GiL Omenn 0 0 3 1 2 8 18 Andrey Archakov 0 0 1 0 1 3 19 20 Siqi Liu 1 0 1 21 Albert Sickmann 0 0 0 0 0 22 X Tadashi Yamamoto 1 0 1 0 Y Hosseini SaLekdeh 1 2 1 1 0 Mt TOTAL 15 268 19 6 13 20 (in progress) 37 C-HPP 2017-09-16 1 The neXt-50 Challenge Chromosome 1 (Ping Xu) PIC Leaders: Ping Xu, Fuchu He Contributing labs: Ping Xu, Beijing Proteome Research Center Fuchu He, Beijing Proteome Research Center Dong Yang, Beijing Proteome Research Center Wantao Ying, Beijing Proteome Research Center Pengyuan Yang, Fudan University Siqi Liu, Beijing Genome Institute Qinyu He, Jinan University Major lab members or partners contributing to the neXt50: Yao Zhang (Beijing Proteome Research Center), Yihao Wang (Beijing Proteome Research Center), Cuitong He (Beijing Proteome Research Center), Wei Wei (Beijing Proteome Research Center), Yanchang Li (Beijing Proteome Research Center), Feng Xu (Beijing Proteome Research Center), Xuehui Peng (Beijing Proteome Research Center). -

CLC and IFNAR1 Are Differentially Expressed and a Global Immunity Score Is Distinct Between Early- and Late-Onset Colorectal Cancer
Genes and Immunity (2011) 12, 653–662 & 2011 Macmillan Publishers Limited All rights reserved 1466-4879/11 www.nature.com/gene ORIGINAL ARTICLE CLC and IFNAR1 are differentially expressed and a global immunity score is distinct between early- and late-onset colorectal cancer TH A˚ gesen1,2, M Berg1,2, T Clancy3, E Thiis-Evensen4, L Cekaite1,2, GE Lind1,2, JM Nesland5,6, A Bakka7, T Mala8, HJ Hauss9, T Fetveit10, MH Vatn4,11, E Hovig3,12,13, A Nesbakken2,6,8, RA Lothe1,2,6 and RI Skotheim1,2 1Department of Cancer Prevention, Institute for Cancer Research, The Norwegian Radium Hospital, Oslo University Hospital, Oslo, Norway; 2Centre for Cancer Biomedicine, University of Oslo, Oslo, Norway; 3Department of Tumor Biology, Institute for Cancer Research, The Norwegian Radium Hospital, Oslo University Hospital, Oslo, Norway; 4Department for Organ Transplantation, Gastroenterology and Nephrology, Rikshospitalet, Oslo University Hospital, Oslo, Norway; 5Division of Pathology, The Norwegian Radium Hospital, Oslo University Hospital, Oslo, Norway; 6Faculty of Medicine, The University of Oslo, Oslo, Norway; 7Department of Digestive Surgery, Akershus University Hospital, Lørenskog, Norway; 8Department of Gastrointestinal Surgery, Aker, Oslo University Hospital, Oslo, Norway; 9Department of Gastrointestinal Surgery, Sørlandet Hospital, Kristiansand, Norway; 10Department of Surgery, Sørlandet Hospital, Arendal, Norway; 11Epigen, Akershus University Hospital, Lørenskog, Norway; 12Institute of Medical Informatics, The Norwegian Radium Hospital, Oslo University -

Understanding Nucleotide Excision Repair and Its Roles in Cancer and Ageing
REVIEWS DNA DAMAGE Understanding nucleotide excision repair and its roles in cancer and ageing Jurgen A. Marteijn*, Hannes Lans*, Wim Vermeulen, Jan H. J. Hoeijmakers Abstract | Nucleotide excision repair (NER) eliminates various structurally unrelated DNA lesions by a multiwise ‘cut and patch’-type reaction. The global genome NER (GG‑NER) subpathway prevents mutagenesis by probing the genome for helix-distorting lesions, whereas transcription-coupled NER (TC‑NER) removes transcription-blocking lesions to permit unperturbed gene expression, thereby preventing cell death. Consequently, defects in GG‑NER result in cancer predisposition, whereas defects in TC‑NER cause a variety of diseases ranging from ultraviolet radiation‑sensitive syndrome to severe premature ageing conditions such as Cockayne syndrome. Recent studies have uncovered new aspects of DNA-damage detection by NER, how NER is regulated by extensive post-translational modifications, and the dynamic chromatin interactions that control its efficiency. Based on these findings, a mechanistic model is proposed that explains the complex genotype–phenotype correlations of transcription-coupled repair disorders. The integrity of DNA is constantly threatened by endo of an intricate DNA-damage response (DDR), which genously formed metabolic products and by-products, comprises sophisticated repair and damage signalling such as reactive oxygen species (ROS) and alkylating processes. The DDR involves DNA-damage sensors and agents, and by its intrinsic chemical instability (for exam signalling kinases that regulate a range of downstream ple, by its ability to spontaneously undergo hydrolytic mediator and effector molecules that control repair, cell deamination and depurination). Environmental chemi cycle progression and cell fate4. The core of this DDR is cals and radiation also affect the physical constitution of formed by a network of complementary DNA repair sys DNA1. -

Genetic Variants in DNA Repair Genes As Potential Predictive Markers for Oxaliplatin Chemotherapy in Colorectal Cancer
The Pharmacogenomics Journal (2015) 15, 505–512 © 2015 Macmillan Publishers Limited All rights reserved 1470-269X/15 www.nature.com/tpj ORIGINAL ARTICLE Genetic variants in DNA repair genes as potential predictive markers for oxaliplatin chemotherapy in colorectal cancer EJ Kap1, P Seibold1, S Richter1, D Scherer2, N Habermann2, Y Balavarca2, L Jansen3, N Becker4, K Pfütze5,6, O Popanda7, M Hoffmeister3, A Ulrich8, A Benner4, CM Ulrich2,9, B Burwinkel5,6, H Brenner3,10 and J Chang-Claude1 Oxaliplatin-based chemotherapy exerts its effects through generating DNA damage. Hence, genetic variants in DNA repair pathways could modulate treatment response. We used a prospective cohort of 623 colorectal cancer patients with stage II–IV disease treated with adjuvant/palliative chemotherapy to comprehensively investigate 1727 genetic variants in the DNA repair pathways as potential predictive markers for oxaliplatin treatment. Single nucleotide polymorphisms (SNP) associations with overall survival and recurrence-free survival were assessed using a Cox regression model. Pathway analysis was performed using the gamma method. Patients carrying variant alleles of rs3783819 (MNAT1) and rs1043953 (XPC) experienced a longer overall survival after treatment with oxaliplatin than patients who did not carry the variant allele, while the opposite association was found in patients who were not treated with oxaliplatin (false discovery rate-adjusted P-values for heterogeneity 0.0047 and 0.0237, respectively). The nucleotide excision repair (NER) pathway was found to be most likely associated with overall survival in patients who received oxaliplatin (P-value = 0.002). Our data show that genetic variants in the NER pathway are potentially predictive of treatment response to oxaliplatin. -

A Software for Detection of Alternative Splicing Events in Human
www.nature.com/scientificreports OPEN SpliceDetector: a software for detection of alternative splicing events in human and model Received: 21 June 2017 Accepted: 2 March 2018 organisms directly from transcript Published: xx xx xxxx IDs Mandana Baharlou Houreh1, Payam Ghorbani Kalkhajeh2, Ali Niazi1, Faezeh Ebrahimi3 & Esmaeil Ebrahimie 1,4,5,6 In eukaryotes, diferent combinations of exons lead to multiple transcripts with various functions in protein level, in a process called alternative splicing (AS). Unfolding the complexity of functional genomics through genome-wide profling of AS and determining the altered ultimate products provide new insights for better understanding of many biological processes, disease progress as well as drug development programs to target harmful splicing variants. The current available tools of alternative splicing work with raw data and include heavy computation. In particular, there is a shortcoming in tools to discover AS events directly from transcripts. Here, we developed a Windows-based user-friendly tool for identifying AS events from transcripts without the need to any advanced computer skill or database download. Meanwhile, due to online working mode, our application employs the updated SpliceGraphs without the need to any resource updating. First, SpliceGraph forms based on the frequency of active splice sites in pre-mRNA. Then, the presented approach compares query transcript exons to SpliceGraph exons. The tool provides the possibility of statistical analysis of AS events as well as AS visualization compared to SpliceGraph. The developed application works for transcript sets in human and model organisms. Transcripts are products of pre-mRNA splicing processes. Novel transcripts discover each day1,2 and add to public databases. -

The P53mh Algorithm and Its Application in Detecting P53-Responsive Genes
The p53MH algorithm and its application in detecting p53-responsive genes J. Hoh*†‡, S. Jin§†, T. Parrado*, J. Edington*, A. J. Levine§, and J. Ott* *Laboratories of Statistical Genetics and §Cancer Biology of The Rockefeller University, 1230 York Avenue, New York, NY 10021 Contributed by A. J. Levine, May 6, 2002 A computer algorithm, p53MH, was developed, which identifies overall binding likelihood is needed for a given gene of arbitrary putative p53 transcription factor DNA-binding sites on a genome- size. Three features have been implemented in an algorithm to wide scale with high power and versatility. With the sequences meet the above requirements: binding propensity plots, weighted from the human and mouse genomes, putative p53 DNA-binding scores, and statistical significance for most likely binding sites. elements were identified in a scan of 2,583 human genes and 1,713 This computer algorithm has been used to identify putative mouse orthologs based on the experimental data of el-Deiry et al. binding elements on a genomewide scale. The algorithm, [el-Deiry, W. S., Kern, S. E., Pietenpol, J. A., Kinzler, K. W. & p53MH, uses an optimal scoring system as an indication of the Vogelstein, B. (1992) Nat. Genet. 1, 45–49] and Funk et al. [Funk, percent similarity to the consensus. It can simultaneously screen W. D., Pak, D. T., Karas, R. H., Wright, W. E. & Shay, J. W. (1992) Mol. thousands of genes for degenerate consensus sequences in the Cell. Biol. 12, 2866–2871] (http:͞͞linkage.rockefeller.edu͞p53). The course of only a few minutes. With this, based on the available p53 DNA-binding motif consists of a 10-bp palindrome and most annotated human sequence databases, a White Page-like direc- commonly a second related palindrome linked by a spacer region. -

Informatics and Clinical Genome Sequencing: Opening the Black Box
©American College of Medical Genetics and Genomics REVIEW Informatics and clinical genome sequencing: opening the black box Sowmiya Moorthie, PhD1, Alison Hall, MA 1 and Caroline F. Wright, PhD1,2 Adoption of whole-genome sequencing as a routine biomedical tool present an overview of the data analysis and interpretation pipeline, is dependent not only on the availability of new high-throughput se- the wider informatics needs, and some of the relevant ethical and quencing technologies, but also on the concomitant development of legal issues. methods and tools for data collection, analysis, and interpretation. Genet Med 2013:15(3):165–171 It would also be enormously facilitated by the development of deci- sion support systems for clinicians and consideration of how such Key Words: bioinformatics; data analysis; massively parallel; information can best be incorporated into care pathways. Here we next-generation sequencing INTRODUCTION Each of these steps requires purpose-built databases, algo- Technological advances have resulted in a dramatic fall in the rithms, software, and expertise to perform. By and large, issues cost of human genome sequencing. However, the sequencing related to primary analysis have been solved and are becom- assay is only the beginning of the process of converting a sample ing increasingly automated, and are therefore not discussed of DNA into meaningful genetic information. The next step of further here. Secondary analysis is also becoming increasingly data collection and analysis involves extensive use of various automated for human genome resequencing, and methods of computational methods for converting raw data into sequence mapping reads to the most recent human genome reference information, and the application of bioinformatics techniques sequence (GRCh37), and calling variants from it, are becom- for the interpretation of that sequence.