Comparative Genomics of the Archaea (Euryarchaeota): Evolution of Conserved Protein Families, the Stable Core, and the Variable Shell
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
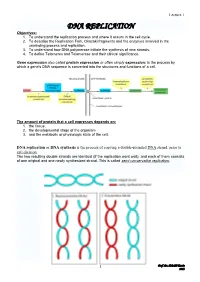
DNA REPLICATION Objectives: 1
Lecture 1 DNA REPLICATION Objectives: 1. To understand the replication process and where it occurs in the cell cycle. 2. To describe the Replication Fork, Okazaki fragments and the enzymes involved in the unwinding process and replication. 3. To understand how DNA polymerase initiate the synthesis of new strands. 4. To define Telomeres and Telomerase and their clinical significance. Gene expression also called protein expression or often simply expression: is the process by which a gene's DNA sequence is converted into the structures and functions of a cell. The amount of protein that a cell expresses depends on: 1. the tissue, 2. the developmental stage of the organism 3. and the metabolic or physiologic state of the cell. DNA replication or DNA synthesis is the process of copying a double-stranded DNA strand, prior to cell division. The two resulting double strands are identical (if the replication went well), and each of them consists of one original and one newly synthesized strand. This is called semi conservative replication. 1 Prof. Dr. H.D.El-Yassin 2013 Lecture 1 The process of replication consists of three steps, initiation, replication and termination. 1. Prokaryotic replication Basic Requirement for DNA Synthesis 1. Substrates: the four deoxy nucleosides triphosphates are needed as substrates for DNA synthesis. Cleavage of the high-energy phosphate bond between the α and β phosphates provides the energy for the addition of the nucleotide. 2. Template: DNA replication cannot occur without a template. A template is required to direct the addition of the appropriate complementary deoxynucleotide to the newly synthesized DNA strand. -
DNA Polymerase Exchange and Lesion Bypass in Escherichia Coli
DNA Polymerase Exchange and Lesion Bypass in Escherichia Coli The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Kath, James Evon. 2016. DNA Polymerase Exchange and Lesion Bypass in Escherichia Coli. Doctoral dissertation, Harvard University, Graduate School of Arts & Sciences. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:26718716 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA ! ! ! ! ! ! ! DNA!polymerase!exchange!and!lesion!bypass!in!Escherichia)coli! ! A!dissertation!presented! by! James!Evon!Kath! to! The!Committee!on!Higher!Degrees!in!Biophysics! ! in!partial!fulfillment!of!the!requirements! for!the!degree!of! Doctor!of!Philosophy! in!the!subject!of! Biophysics! ! Harvard!University! Cambridge,!Massachusetts! October!2015! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ©!2015!L!James!E.!Kath.!Some!Rights!Reserved.! ! This!work!is!licensed!under!the!Creative!Commons!Attribution!3.0!United!States!License.!To! view!a!copy!of!this!license,!visit:!http://creativecommons.org/licenses/By/3.0/us! ! ! Dissertation!Advisor:!Professor!Joseph!J.!Loparo! ! ! !!!!!!!!James!Evon!Kath! ! DNA$polymerase$exchange$and$lesion$bypass$in$Escherichia)coli$ $ Abstract$ ! Translesion! synthesis! (TLS)! alleviates! -

Methane Production on Rock and Soil Substrates by Methanogens: Implications for Life on Mars
Bioastronomy 2007: Molecules, Microbes, and Extraterrestrial Life ASP Conference Series, Vol. 420, 2009 K. J. Meech, J. V. Keane, M. J. Mumma, J. L. Siefert, and D. J. Werthimer, eds. Methane Production on Rock and Soil Substrates by Methanogens: Implications for Life on Mars H. A. Kozup and T. A. Kral 1West Virginia University, WV 26505 2Arkansas Center for Space and Planetary Sciences, University of Arkansas, Fayetteville 72701 Abstract. In order to understand the methanogens as models for possible life on Mars, and some of the factors likely to be important in determining their abundance and distribution, we have measured their ability to produce methane on a few types of inorganic rock and soil substrates. Since organic ma- terials have not been detected in measurable quantities at the surface of Mars, there is no reason to believe that they would exist in the subsurface. Samples of three methanogens (Methanosarcina barkeri, Methanobacterium formicicum, and Methanothermobacter wolfeii) were placed on four substrates (sand, gravel, basalt, and a Mars soil simulant, JSC Mars-1) and methane production mea- sured. Glass beads were used as a control substrate. As in earlier experiments with JSC Mars-1 soil simulant, a crushed volcanic tephra, methane was pro- duced by all three methanogens when placed on the substrates, sand and gravel. None produced methane on basalt in these experiments, a mineral common in Martian soil. While these substrates do not represent the full range of materials likely to be present on the surface of Mars, the present results suggest that while some surface materials on Mars may not support this type of organism, others might. -

A Web Tool for Hydrogenase Classification and Analysis Dan Søndergaard1, Christian N
www.nature.com/scientificreports OPEN HydDB: A web tool for hydrogenase classification and analysis Dan Søndergaard1, Christian N. S. Pedersen1 & Chris Greening2,3 H2 metabolism is proposed to be the most ancient and diverse mechanism of energy-conservation. The Received: 24 June 2016 metalloenzymes mediating this metabolism, hydrogenases, are encoded by over 60 microbial phyla Accepted: 09 September 2016 and are present in all major ecosystems. We developed a classification system and web tool, HydDB, Published: 27 September 2016 for the structural and functional analysis of these enzymes. We show that hydrogenase function can be predicted by primary sequence alone using an expanded classification scheme (comprising 29 [NiFe], 8 [FeFe], and 1 [Fe] hydrogenase classes) that defines 11 new classes with distinct biological functions. Using this scheme, we built a web tool that rapidly and reliably classifies hydrogenase primary sequences using a combination of k-nearest neighbors’ algorithms and CDD referencing. Demonstrating its capacity, the tool reliably predicted hydrogenase content and function in 12 newly-sequenced bacteria, archaea, and eukaryotes. HydDB provides the capacity to browse the amino acid sequences of 3248 annotated hydrogenase catalytic subunits and also contains a detailed repository of physiological, biochemical, and structural information about the 38 hydrogenase classes defined here. The database and classifier are freely and publicly available at http://services.birc.au.dk/hyddb/ Microorganisms conserve energy by metabolizing H2. Oxidation of this high-energy fuel yields electrons that can be used for respiration and carbon-fixation. This diffusible gas is also produced in diverse fermentation and 1 anaerobic respiratory processes . H2 metabolism contributes to the growth and survival of microorganisms across the three domains of life, including chemotrophs and phototrophs, lithotrophs and heterotrophs, aerobes and 1,2 anaerobes, mesophiles and extremophiles alike . -

Ancient Metaproteomics: a Novel Approach for Understanding Disease And
Ancient metaproteomics: a novel approach for understanding disease and diet in the archaeological record Jessica Hendy PhD University of York Archaeology August, 2015 ii Abstract Proteomics is increasingly being applied to archaeological samples following technological developments in mass spectrometry. This thesis explores how these developments may contribute to the characterisation of disease and diet in the archaeological record. This thesis has a three-fold aim; a) to evaluate the potential of shotgun proteomics as a method for characterising ancient disease, b) to develop the metaproteomic analysis of dental calculus as a tool for understanding both ancient oral health and patterns of individual food consumption and c) to apply these methodological developments to understanding individual lifeways of people enslaved during the 19th century transatlantic slave trade. This thesis demonstrates that ancient metaproteomics can be a powerful tool for identifying microorganisms in the archaeological record, characterising the functional profile of ancient proteomes and accessing individual patterns of food consumption with high taxonomic specificity. In particular, analysis of dental calculus may be an extremely valuable tool for understanding the aetiology of past oral diseases. Results of this study highlight the value of revisiting previous studies with more recent methodological approaches and demonstrate that biomolecular preservation can have a significant impact on the effectiveness of ancient proteins as an archaeological tool for this characterisation. Using the approaches developed in this study we have the opportunity to increase the visibility of past diseases and their aetiology, as well as develop a richer understanding of individual lifeways through the production of molecular life histories. iii iv List of Contents Abstract ............................................................................................................................... -

Molecular Insights Into DNA Interference by CRISPR-Associated Nuclease-Helicase Cas3
Molecular insights into DNA interference by CRISPR-associated nuclease-helicase Cas3 Bei Gonga,1, Minsang Shinb,1, Jiali Suna, Che-Hun Junga,b, Edward L. Boltc, John van der Oostd, and Jeong-Sun Kima,b,2 aInterdisciplinary Graduate Program in Molecular Medicine, Chonnam National University, Gwangju 501-746, Korea; bDepartment of Chemistry, Chonnam National University, Gwangju 500-757, Korea; cSchool of Life Sciences, University of Nottingham Medical School, Nottingham NG72UH, United Kingdom; and dLaboratory of Microbiology, Wageningen University, 6703 HB, Wageningen, The Netherlands Edited by Wei Yang, National Institutes of Health, Bethesda, MD, and approved September 26, 2014 (received for review June 10, 2014) Mobile genetic elements in bacteria are neutralized by a system 21), Cascade complex by itself is not a nuclease for degradation based on clustered regularly interspaced short palindromic repeats of invader DNA (13). In Escherichia coli K-12 (type IE), Cascade (CRISPRs) and CRISPR-associated (Cas) proteins. Type I CRISPR-Cas recognizes and binds crRNA to complimentary sequence in systems use a “Cascade” ribonucleoprotein complex to guide RNA target DNA, generating an RNA mediated displacement loop specifically to complementary sequence in invader double-stranded (R-loop) of single-stranded (ss) DNA and RNA-DNA hybrid DNA (dsDNA), a process called “interference.” After target recogni- within double-stranded (ds) target DNA (13). Formation of the tion by Cascade, formation of an R-loop triggers recruitment of R-loop structure induces conformational change in Cascade subunits, triggering recruitment of Cas3 protein that is a nucle- a Cas3 nuclease-helicase, completing the interference process by – destroying the invader dsDNA. -

Part One Amino Acids As Building Blocks
Part One Amino Acids as Building Blocks Amino Acids, Peptides and Proteins in Organic Chemistry. Vol.3 – Building Blocks, Catalysis and Coupling Chemistry. Edited by Andrew B. Hughes Copyright Ó 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim ISBN: 978-3-527-32102-5 j3 1 Amino Acid Biosynthesis Emily J. Parker and Andrew J. Pratt 1.1 Introduction The ribosomal synthesis of proteins utilizes a family of 20 a-amino acids that are universally coded by the translation machinery; in addition, two further a-amino acids, selenocysteine and pyrrolysine, are now believed to be incorporated into proteins via ribosomal synthesis in some organisms. More than 300 other amino acid residues have been identified in proteins, but most are of restricted distribution and produced via post-translational modification of the ubiquitous protein amino acids [1]. The ribosomally encoded a-amino acids described here ultimately derive from a-keto acids by a process corresponding to reductive amination. The most important biosynthetic distinction relates to whether appropriate carbon skeletons are pre-existing in basic metabolism or whether they have to be synthesized de novo and this division underpins the structure of this chapter. There are a small number of a-keto acids ubiquitously found in core metabolism, notably pyruvate (and a related 3-phosphoglycerate derivative from glycolysis), together with two components of the tricarboxylic acid cycle (TCA), oxaloacetate and a-ketoglutarate (a-KG). These building blocks ultimately provide the carbon skeletons for unbranched a-amino acids of three, four, and five carbons, respectively. a-Amino acids with shorter (glycine) or longer (lysine and pyrrolysine) straight chains are made by alternative pathways depending on the available raw materials. -

A Proposal to Rename the Hyperthermophile Pyrococcus Woesei As Pyrococcus Furiosus Subsp
Archaea 1, 277–283 © 2004 Heron Publishing—Victoria, Canada A proposal to rename the hyperthermophile Pyrococcus woesei as Pyrococcus furiosus subsp. woesei WIROJNE KANOKSILAPATHAM,1 JUAN M. GONZÁLEZ,1,2 DENNIS L. MAEDER,1 1,3 1,4 JOCELYNE DIRUGGIERO and FRANK T. ROBB 1 Center of Marine Biotechnology, University of Maryland Biotechnology Institute, Baltimore, MD 21202, USA 2 Present address: IRNAS-CSIC, P.O. Box 1052, 41080 Sevilla, Spain 3 Department of Cell Biology and Molecular Genetics, University of Maryland, College Park, MD 20274, USA 4 Corresponding author ([email protected]) Received April 8, 2004; accepted July 28, 2004; published online August 31, 2004 Summary Pyrococcus species are hyperthermophilic mem- relatively rapidly at temperatures above 90 °C and produce ° bers of the order Thermococcales, with optimal growth tem- H2S from elemental sulfur (S ) (Zillig et al. 1987). Two Pyro- peratures approaching 100 °C. All species grow heterotrophic- coccus species, P. furiosus (Fiala and Stetter 1986) and ° ally and produce H2 or, in the presence of elemental sulfur (S ), P. woesei (Zillig et al. 1987), were isolated from marine sedi- H2S. Pyrococcus woesei and P.furiosus were isolated from ma- ments at a Vulcano Island beach site in Italy. They have similar rine sediments at the same Vulcano Island beach site and share morphological and physiological characteristics: both species many morphological and physiological characteristics. We re- are cocci and move by means of a tuft of polar flagella. They port here that the rDNA operons of these strains have identical are heterotrophic and grow optimally at 95–100 °C, utilizing sequences, including their intergenic spacer regions and part of peptides as major carbon and nitrogen sources. -

Histone Variants in Archaea and the Evolution of Combinatorial Chromatin Complexity
Histone variants in archaea and the evolution of combinatorial chromatin complexity Kathryn M. Stevensa,b, Jacob B. Swadlinga,b, Antoine Hochera,b, Corinna Bangc,d, Simonetta Gribaldoe, Ruth A. Schmitzc, and Tobias Warneckea,b,1 aMolecular Systems Group, Quantitative Biology Section, Medical Research Council London Institute of Medical Sciences, London W12 0NN, United Kingdom; bInstitute of Clinical Sciences, Faculty of Medicine, Imperial College London, London W12 0NN, United Kingdom; cInstitute for General Microbiology, University of Kiel, 24118 Kiel, Germany; dInstitute of Clinical Molecular Biology, University of Kiel, 24105 Kiel, Germany; and eDepartment of Microbiology, Unit “Evolutionary Biology of the Microbial Cell,” Institut Pasteur, 75015 Paris, France Edited by W. Ford Doolittle, Dalhousie University, Halifax, NS, Canada, and approved October 28, 2020 (received for review April 14, 2020) Nucleosomes in eukaryotes act as platforms for the dynamic inte- additional histone dimers can be taggedontothistetramertoyield gration of epigenetic information. Posttranslational modifications oligomers of increasing length that wrap correspondingly more DNA are reversibly added or removed and core histones exchanged for (3, 6–9). Almost all archaeal histones lack tails and PTMs have yet to paralogous variants, in concert with changing demands on tran- be reported. Many archaea do, however, encode multiple histone scription and genome accessibility. Histones are also common in paralogs (8, 10) that can flexibly homo- and heterodimerize in -

Archaeoglobus Profundus Type Strain (AV18T)
Standards in Genomic Sciences (2010) 2:327-346 DOI:10.4056/sigs.942153 Complete genome sequence of Archaeoglobus profundus type strain (AV18T) Mathias von Jan1, Alla Lapidus2, Tijana Glavina Del Rio2, Alex Copeland2, Hope Tice2, Jan-Fang Cheng2, Susan Lucas2, Feng Chen2, Matt Nolan2, Lynne Goodwin2,3, Cliff Han2,3, Sam Pitluck2, Konstantinos Liolios2, Natalia Ivanova2, Konstantinos Mavromatis2, Galina Ovchinnikova2, Olga Chertkov2, Amrita Pati2, Amy Chen4, Krishna Palaniappan4, Miriam Land2,5, Loren Hauser2,5, Yun-Juan Chang2,5, Cynthia D. Jeffries2,5, Elizabeth Saunders2, Thomas Brettin2,3, John C. Detter2,3, Patrick Chain2,4, Konrad Eichinger6, Harald Huber6, Ste- fan Spring1, Manfred Rohde7, Markus Göker1, Reinhard Wirth6, Tanja Woyke2, Jim Bristow2, Jonathan A. Eisen2,8, Victor Markowitz4, Philip Hugenholtz2, Nikos C Kyrpides2, and Hans-Peter Klenk1* 1 DSMZ - German Collection of Microorganisms and Cell Cultures GmbH, Braunschweig, Germany 2 DOE Joint Genome Institute, Walnut Creek, California, USA 3 Los Alamos National Laboratory, Bioscience Division, Los Alamos, New Mexico, USA 4 Biological Data Management and Technology Center, Lawrence Berkeley National Laboratory, Berkeley, California, USA 5 Oak Ridge National Laboratory, Oak Ridge, Tennessee, USA 6 University of Regensburg, Microbiology – Archaeenzentrum, Regensburg, Germany 7 HZI – Helmholtz Centre for Infection Research, Braunschweig, Germany 8 University of California Davis Genome Center, Davis, California, USA *Corresponding author: Hans-Peter Klenk Keywords: hyperthermophilic, marine, strictly anaerobic, sulfate respiration, hydrogen utili- zation, hydrothermal systems, Archaeoglobaceae, GEBA Archaeoglobus profundus (Burggraf et al. 1990) is a hyperthermophilic archaeon in the eu- ryarchaeal class Archaeoglobi, which is currently represented by the single family Archaeog- lobaceae, containing six validly named species and two strains ascribed to the genus 'Geoglobus' which is taxonomically challenged as the corresponding type species has no va- lidly published name. -

Genomic Analysis of Family UBA6911 (Group 18 Acidobacteria)
bioRxiv preprint doi: https://doi.org/10.1101/2021.04.09.439258; this version posted April 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 2 Genomic analysis of family UBA6911 (Group 18 3 Acidobacteria) expands the metabolic capacities of the 4 phylum and highlights adaptations to terrestrial habitats. 5 6 Archana Yadav1, Jenna C. Borrelli1, Mostafa S. Elshahed1, and Noha H. Youssef1* 7 8 1Department of Microbiology and Molecular Genetics, Oklahoma State University, Stillwater, 9 OK 10 *Correspondence: Noha H. Youssef: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2021.04.09.439258; this version posted April 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 11 Abstract 12 Approaches for recovering and analyzing genomes belonging to novel, hitherto unexplored 13 bacterial lineages have provided invaluable insights into the metabolic capabilities and 14 ecological roles of yet-uncultured taxa. The phylum Acidobacteria is one of the most prevalent 15 and ecologically successful lineages on earth yet, currently, multiple lineages within this phylum 16 remain unexplored. Here, we utilize genomes recovered from Zodletone spring, an anaerobic 17 sulfide and sulfur-rich spring in southwestern Oklahoma, as well as from multiple disparate soil 18 and non-soil habitats, to examine the metabolic capabilities and ecological role of members of 19 the family UBA6911 (group18) Acidobacteria. -

Yeast Genome Gazetteer P35-65
gazetteer Metabolism 35 tRNA modification mitochondrial transport amino-acid metabolism other tRNA-transcription activities vesicular transport (Golgi network, etc.) nitrogen and sulphur metabolism mRNA synthesis peroxisomal transport nucleotide metabolism mRNA processing (splicing) vacuolar transport phosphate metabolism mRNA processing (5’-end, 3’-end processing extracellular transport carbohydrate metabolism and mRNA degradation) cellular import lipid, fatty-acid and sterol metabolism other mRNA-transcription activities other intracellular-transport activities biosynthesis of vitamins, cofactors and RNA transport prosthetic groups other transcription activities Cellular organization and biogenesis 54 ionic homeostasis organization and biogenesis of cell wall and Protein synthesis 48 plasma membrane Energy 40 ribosomal proteins organization and biogenesis of glycolysis translation (initiation,elongation and cytoskeleton gluconeogenesis termination) organization and biogenesis of endoplasmic pentose-phosphate pathway translational control reticulum and Golgi tricarboxylic-acid pathway tRNA synthetases organization and biogenesis of chromosome respiration other protein-synthesis activities structure fermentation mitochondrial organization and biogenesis metabolism of energy reserves (glycogen Protein destination 49 peroxisomal organization and biogenesis and trehalose) protein folding and stabilization endosomal organization and biogenesis other energy-generation activities protein targeting, sorting and translocation vacuolar and lysosomal