Summary of Product Characteristics 1. Name Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Misuse of Benzodiazepines Among High-Risk Opioid Users in Europe
EMBARGO — 7 JUNE 7. 6. 2018 UPDATED 11:30 Central European Time/CET (10:30 Western European Time/WET/Lisbon) Proof - 28 May 2018 not for circulation PERSPECTIVES ON DRUGS The misuse of benzodiazepines among high-risk opioid users in Europe Benzodiazepines are a widely prescribed I Introduction group of medicines with a range of clinical uses that include treating Benzodiazepines have a range of clinical uses and are among the most commonly prescribed medicines globally. anxiety, insomnia and managing alcohol They are useful in the short-term treatment of anxiety and withdrawal. This group of medicines is insomnia, and in managing alcohol withdrawal (Medicines often misused by high-risk opioid users, and Healthcare Products Regulatory Agency, 2015). Like all medicines, benzodiazepines can produce side effects. They and this is associated with considerable may also be misused, which we define as use without a morbidity and mortality. This paper prescription from a medical practitioner or, if prescribed, when describes the impact of benzodiazepines they are used outside accepted medical practice or guidelines. misuse on the health and treatment of While the misuse of benzodiazepines has been identified high-risk opioid users. as a concern for large groups in the general population, for example, among elderly people and women, this analysis focuses specifically on misuse among high-risk opioid users (1), a group of people among whom these medicines have been linked with severe treatment challenges and implicated in considerable numbers of drug-related deaths. It is important to stress that much benzodiazepine prescribing to high-risk drug users is done with legitimate therapeutic aims in mind. -

Drugs of Abuse: Benzodiazepines
Drugs of Abuse: Benzodiazepines What are Benzodiazepines? Benzodiazepines are central nervous system depressants that produce sedation, induce sleep, relieve anxiety and muscle spasms, and prevent seizures. What is their origin? Benzodiazepines are only legally available through prescription. Many abusers maintain their drug supply by getting prescriptions from several doctors, forging prescriptions, or buying them illicitly. Alprazolam and diazepam are the two most frequently encountered benzodiazepines on the illicit market. Benzodiazepines are What are common street names? depressants legally available Common street names include Benzos and Downers. through prescription. Abuse is associated with What do they look like? adolescents and young The most common benzodiazepines are the prescription drugs ® ® ® ® ® adults who take the drug Valium , Xanax , Halcion , Ativan , and Klonopin . Tolerance can orally or crush it up and develop, although at variable rates and to different degrees. short it to get high. Shorter-acting benzodiazepines used to manage insomnia include estazolam (ProSom®), flurazepam (Dalmane®), temazepam (Restoril®), Benzodiazepines slow down and triazolam (Halcion®). Midazolam (Versed®), a short-acting the central nervous system. benzodiazepine, is utilized for sedation, anxiety, and amnesia in critical Overdose effects include care settings and prior to anesthesia. It is available in the United States shallow respiration, clammy as an injectable preparation and as a syrup (primarily for pediatric skin, dilated pupils, weak patients). and rapid pulse, coma, and possible death. Benzodiazepines with a longer duration of action are utilized to treat insomnia in patients with daytime anxiety. These benzodiazepines include alprazolam (Xanax®), chlordiazepoxide (Librium®), clorazepate (Tranxene®), diazepam (Valium®), halazepam (Paxipam®), lorzepam (Ativan®), oxazepam (Serax®), prazepam (Centrax®), and quazepam (Doral®). -

The Emergence of New Psychoactive Substance (NPS) Benzodiazepines
Issue: Ir Med J; Vol 112; No. 7; P970 The Emergence of New Psychoactive Substance (NPS) Benzodiazepines. A Survey of their Prevalence in Opioid Substitution Patients using LC-MS S. Mc Namara, S. Stokes, J. Nolan HSE National Drug Treatment Centre Abstract Benzodiazepines have a wide range of clinical uses being among the most commonly prescribed medicines globally. The EU Early Warning System on new psychoactive substances (NPS) has over recent years detected new illicit benzodiazepines in Europe’s drug market1. Additional reference standards were obtained and a multi-residue LC- MS method was developed to test for 31 benzodiazepines or metabolites in urine including some new benzodiazepines which have been classified as New Psychoactive Substances (NPS) which comprise a range of substances, including synthetic cannabinoids, opioids, cathinones and benzodiazepines not covered by international drug controls. 200 urine samples from patients attending the HSE National Drug Treatment Centre (NDTC) who are monitored on a regular basis for drug and alcohol use and which tested positive for benzodiazepine class drugs by immunoassay screening were subjected to confirmatory analysis to determine what Benzodiazepine drugs were present and to see if etizolam or other new benzodiazepines are being used in the addiction population currently. Benzodiazepine prescription and use is common in the addiction population. Of significance we found evidence of consumption of an illicit new psychoactive benzodiazepine, Etizolam. Introduction Benzodiazepines are useful in the short-term treatment of anxiety and insomnia, and in managing alcohol withdrawal. 1 According to the EMCDDA report on the misuse of benzodiazepines among high-risk opioid users in Europe1, benzodiazepines, especially when injected, can prolong the intensity and duration of opioid effects. -

A Review of the Evidence of Use and Harms of Novel Benzodiazepines
ACMD Advisory Council on the Misuse of Drugs Novel Benzodiazepines A review of the evidence of use and harms of Novel Benzodiazepines April 2020 1 Contents 1. Introduction ................................................................................................................................. 4 2. Legal control of benzodiazepines .......................................................................................... 4 3. Benzodiazepine chemistry and pharmacology .................................................................. 6 4. Benzodiazepine misuse............................................................................................................ 7 Benzodiazepine use with opioids ................................................................................................... 9 Social harms of benzodiazepine use .......................................................................................... 10 Suicide ............................................................................................................................................. 11 5. Prevalence and harm summaries of Novel Benzodiazepines ...................................... 11 1. Flualprazolam ......................................................................................................................... 11 2. Norfludiazepam ....................................................................................................................... 13 3. Flunitrazolam .......................................................................................................................... -

Analysis of Benzodiazepines in Blood by LC/MS/MS Application Note
Analysis of Benzodiazepines in Blood by LC/MS/MS Application Note Forensic Toxicology Authors Introduction Hazel Rivera and G. Stewart Walker The analysis of benzodiazepines is of great interest Flinders University to forensic toxicologists. Adelaide, South Australia Australia Screening of these compounds has been problem- atic since immunoassays are often not sufficiently Peter Stockham and D. Noel Sims specific or sensitive enough for low-dosage benzo- Forensic Science South Australia diazepines, especially in blood. Benzodiazepines Adelaide, South Australia have been analyzed using gas chromatography/ Australia nitrogen phosphorus detector (GC/NPD) [1], gas John M. Hughes chromatography/electron capture detector (GC/ECD) [1], and gas chromatography/mass spec- Agilent Technologies, Inc. trometry (GC/MS) [2, 3]. Many benzodiazepines are Pleasanton, CA polar and thermally labile, making them diffi-cult, USA if not impossible, to analyze with GC or GC/MS without derivatization. Some of the com- Abstract pounds cannot be derivatized for improved chromatographic behavior. A sensitive and selective method for the simultaneous screening and identification of 13 benzodiazepines and Screening for benzodiazepines can also be carried 5 metabolites in human blood using the Agilent LC/MSD out using HPLC with UV detection [4], but this Trap is described. The method uses liquid-liquid extrac- technique lacks both the sensitivity and specificity tion followed by reverse-phase LC/MS/MS (liquid required for forensic applications. Furthermore, chromatography/tandem mass spectrometry). The some of the newer benzodiazepines, like fluni- technique is suitable for screening analysis and trazepam, have much lower usage dose ranges and high-confidence identification of the analytes at their faster clearance, and therefore require identifica- lowest reported dosage concentrations using only 500 µL tion at lower levels. -

Drug, Drug Metabolites Trade Names, Street Name
Drug, Drug metabolites Trade names, street name Opioids Methadone Dolophine, Methadose EDDP Buprenorphine Buprenex, Suboxone, Subutex Norbuprenorphine cis-Tramadol Ultram O-DSM-Tramadol Codeine Tylenol3, Tylenol4, Tylenol5, morphine metabolite Morphine Astromorph, Avinza, DepoDur, Duramorph, Kadian, MS Contin; heroin metabolite Fentanyl Actiq, Duragesic, Fentora, Lonsys, Sublimaze Hydrocodone Synalgos_DC, Anexsia, Damason-P, Hycodan, Lortab, Maxidone, Norco, Panacet, Vicodin, Zydone; codeine metabolite Hydromorphone Dilaudid; Hymorphan; morphine and hydrocodone metabolite Meperidine Demerol, Mepergan Naloxone Narcan, Nalone, Evzio, Narcanti Naltrexone Revia, Depade, Vivitrol Oxycodone Oxycontin (with acetaminophen: Endocet, Percocet, Roxicet; with aspirin: Endodan, Percodan, Roxiprin; with ibuprofen: Combunox) Oxymorphone Numorphan, Opana; oxycodone metabolite Stimulants Amphetamine Benzedrine, Dexedrine, Adderall, Vyvanse, speed; could be methamphetamine metabolite; if so, typically < 30 percent of parent Methamphetamine Desoxyn, methedrine, Vicks inhaler (D- and L-isomers are not resolved; low concentrations expected if the source is Vicks); selegeline (Atapryl, Carbex, Carbex, Eldepryl, Zelapar) metabolite MDMA XTC, ecstacy, Adam, bean, , Mercedes, Molly, New Yorkers, peace, roll, white dove, X MDA MDMA and MDEA metabolite 6-MAM Heroin metabolite Benzoylecgonine Cocaine metabolite, Esterom Cocaine Crack; parent drug rarely observed due to short half-life. Phencyclidine PCP, angel dust MDEA Eve Benzodiazepines Alprazolam Xanax alpha-Hydroxyalprazolam -

INFORMATION for the USER PRAZEPAM EG 15 Mg/Ml Oral
PACKAGE LEAFLET: INFORMATION FOR THE USER PRAZEPAM EG 15 mg/ml oral drops, solution prazepam Read all of this leaflet carefully before you start taking this medicine because it contains important information for you. Keep this leaflet. You may need to read it again. If you have any further questions, ask your doctor or pharmacist. This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours. If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet. See section 4. What is in this leaflet 1. What Prazepam EG is and what it is used for 2. What you need to know before you take Prazepam EG 3. How to take Prazepam EG 4. Possible side effects 5. How to store Prazepam EG 6. Contents of the pack and other information 1. What Prazepam EG is and what it is used for The active substance of Prazepam EG is prazepam. Prazepam is a benzodiazepine derivative and is indicated to treat symptoms of anxiety. Benzodiazepines are used to treat severe incapacitating symptoms or symptoms leading to an extreme suffering for the patient. Prazepam EG is used to treat anxiety and nervous tension requiring sedative treatment. 2. What you need to know before you take Prazepam EG DO NOT take Prazepam EG: if you are allergic to prazepam or any of the other ingredients of this medicine (listed in section 6) if you have experienced an allergic reaction to other benzodiazepines in the past if you suffer from -
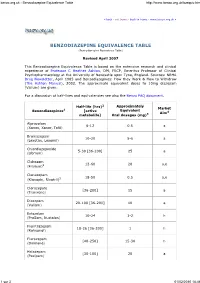
Benzodiazepine Equivalence Table
benzo.org.uk : Benzodiazepine Equivalence Table http://www.benzo.org.uk/bzequiv.htm « back exit frame back to frame www.benzo.org.uk » BENZODIAZEPINE EQUIVALENCE TABLE (Benzodiazepine Equivalency Table) Revised April 2007 This Benzodiazepine Equivalence Table is based on the extensive research and clinical experience of Professor C Heather Ashton , DM, FRCP, Emeritus Professor of Clinical Psychopharmacology at the University of Newcastle upon Tyne, England. Sources: NRHA Drug Newsletter , April 1985 and Benzodiazepines: How they Work & How to Withdraw (The Ashton Manual ), 2002. The approximate equivalent doses to 10mg diazepam (Valium) are given. For a discussion of half-lives and equivalencies see also the Benzo FAQ document . 2 Approximately Half-life (hrs) Market Benzodiazepines 1 [active Equivalent Aim4 metabolite] Oral dosages (mg) 3 Alprazolam 6-12 0.5 a (Xanax, Xanor, Tafil) Bromazepam 10-20 5-6 a (Lexotan, Lexomil) Chlordiazepoxide 5-30 [36-200] 25 a (Librium) Clobazam 12-60 20 a,e (Frisium) 5 Clonazepam 18-50 0.5 a,e (Klonopin, Rivotril) 5 Clorazepate [36-200] 15 a (Tranxene) Diazepam 20-100 [36-200] 10 a (Valium) Estazolam 10-24 1-2 h (ProSom, Nuctalon) Flunitrazepam 18-26 [36-200] 1 h (Rohypnol) Flurazepam [40-250] 15-30 h (Dalmane) Halazepam [30-100] 20 a (Paxipam) 1 sur 3 01/02/2010 18:46 benzo.org.uk : Benzodiazepine Equivalence Table http://www.benzo.org.uk/bzequiv.htm Ketazolam 30-100 [36-200] 15-30 a (Anxon) Loprazolam 6-12 1-2 h (Dormonoct) Lorazepam 10-20 1 a (Ativan, Temesta, Tavor) Lormetazepam 10-12 1-2 h (Noctamid) Medazepam 36-200 10 a (Nobrium) Nitrazepam 15-38 10 h (Mogadon) Nordazepam 36-200 10 a (Nordaz, Calmday) Oxazepam (Serax, Serenid, 4-15 20 a Serepax, Seresta) Prazepam [36-200] 10-20 a (Centrax, Lysanxia) Quazepam (Doral) 25-100 20 h Temazepam (Restoril, Normison, 8-22 20 h Euhypnos) Triazolam 2 0.5 h (Halcion) Non-benzodiazepines with similar effects 1, 6 Zaleplon 2 20 h (Sonata) Zolpidem (Ambien, Stilnoct, 2 20 h Stilnox) Zopiclone 5-6 15 h (Zimovane, Imovane) Eszopiclone 6 3 h (Lunesta) (9 in elderly) 1. -
Forensic Intoxication with Clobazam: HPLC/DAD/MSD Analysis Paula Proenc¸Aa,*, Helena Teixeiraa,B, Joa˜O Pinheiroa, Estela P
Forensic Science International 143 (2004) 205–209 Forensic intoxication with clobazam: HPLC/DAD/MSD analysis Paula Proenc¸aa,*, Helena Teixeiraa,b, Joa˜o Pinheiroa, Estela P. Marquesa,b, Duarte Nuno Vieiraa,b aNational Institute of Legal Medicine—Delegation of Coimbra, Largo da Se´ Nova, 3000-213 Coimbra, Portugal bFaculty of Medicine, University of Coimbra, Largo da Se´ Nova, 3000-213 Coimbra, Portugal Received 20 November 2003; accepted 1 March 2004 Available online 28 May 2004 Abstract Clobazam (Castillium1, Urbanil1), a benzodiazepine often used as an anxiolytic and in the treatment of epilepsy, is considered a relatively safe drug. The authors present a fatal case with a 49-year-old female, found dead at home. She had been undergoing psychiatric treatment and was a chronic alcoholic. The autopsy findings were unremarkable, except for multivisceral congestion, steatosis and a small piece of a plastic blister pack in the stomach. Bronchopneumonia, bronchitis and bronchiolitis were also diagnosed. Anhigh-performance liquid chromatography (HPLC)/diode array detector (DAD)/mass spectrometry detection (MSD) with electrospray method was developed in order to detect, confirm and quantify clobazam in the post-mortem samples. In the chromatographic separation, a reversed-phase column C18 (2:1 Â 150 mm, 3.5 mm) was used with a mobile phase of methanol and water, at a 0.25 ml/min flow rate. Carbonate buffer (pH 10.5) and 20 ml of prazepam (100 mg/ml) as internal standard were added to the samples. A simple and reliable liquid–liquid extraction method for the determination of clobazam in post-mortem samples was described. -

Commonly Prescribed Psychotropic Medications
Commonly Prescribed Psychotropic Medications Antipsychotics (used in the treatment of Anti-depressants Anti-obsessive Agents schizophrenia and mania) Typical Antipsychotics Tricyclics Haldol (haloperidol) *Anafranil (clomipramine) Anafranil (clomipramine) Loxitane (loxapine) Asendin (amoxapine) Luvox (fluvoxamine) Mellaril (thioridazine) Elavil (amitriptyline) Paxil (paroxetine) Moban (molindone) Norpramin (desipramine) Prozac (fluoxetine) Navane (thiothixene) Pamelor (nortriptyline) Zoloft (sertraline) Prolixin (fluphenazine) Sinequan (doxepin) Serentil (mesoridazine) Surmontil (trimipramine) Antianxiety Agents Stelazine (trifluoperazine) Tofranil (imipramine) Ativan (lorazepam) Thorazine (chlorpromazine) Vivactil (protiptyline) BuSpar (buspirone) Trilafon (perphenazine) Centrax (prazepam) SSRIs *Inderal (propranolol) Atypical Antipsychotics Celexa (citalopram) *Klonopin (clonazepam) Abilify (aripiprazole) Lexapro (escitalopram) Lexapro (escitalopram) Clozaril (clozapine) *Luvox (fluvoxamine) Librium (chlordiazepoxide) Risperdal (risperidone) Paxil (paroxetine) Serax (oxazepam) Seroquel (quetiapine) Prozac (fluoxetine) *Tenormin (atenolol) Zyprexa (olanzapine) Zoloft (sertraline) Tranxene (clorazepate) Valium (diazepam) MAOIs Xanax (alprazolam) Mood Stabilizers *Antidepressants, especially Nardil (phenelzine) (used in the treatment SSRIs, are also used in the Parnate (tranylcypromine) of bipolar disorder) treatment of anxiety. Depakene (valproic acid) Depakote Others Stimulants Eskalith Desyrel (trazadone) (used in the Page 1 of 4 Lithobid -

Safe Drug Use for the Recovering Addict Or Alcoholic
Safe Drug Use for the Recovering Addict or Alcoholic 3995 South Cobb Drive Smyrna, Georgia 30080 770-434-4567 1-800-329-9775 www.ridgeviewinstitute.com Revised September 2012 Safe Drug Use for the Recovering Addict or Alcoholic People in recovery must be especially careful when taking any Pain meds kind of over-the-counter (otc) or prescription medications. • Pain meds are tricky for people in recovery. Most otc pain Many otc meds contain alcohol or other ingredients that relievers are fine — ibuprofen (Advil, Motrin), naproxyn could endanger their sobriety by triggering a relapse. Even (Aleve), Tylenol. These meds are very effective for many physicians not familiar with addiction may prescribe meds aches and pains. There are times when the addict/alcoholic that are not safe for the addict/alcoholic. People in recovery must have stronger pain meds, such as narcotics, after must be vigilant in protecting their sobriety. They must read surgery or for a severe injury. If the narcotics are necessary ingredients, ask questions, and use much caution in using any and taken only as directed, this is not considered a relapse. kind of medication. If in doubt about a specific medication, It is normal for this to cause anxiety in recovering people, contact your psychiatrist/addictionologist or another knowl- so it is recommended that the recovering person prepare edgeable person for guidance. themselves before surgery when possible with extra recovery support and pre-planning regarding pain relief. In Important points: any case, the recovering person should not handle the pain • Never take a medication given to you by someone else meds themselves. -

Poisonings and Deaths Caused by Benzodiazepine Drugs in Costa Rica, from 2007 to 2014
MOJ Toxicology Research Article Open Access Poisonings and deaths caused by benzodiazepine drugs in costa rica, from 2007 to 2014 Abstract Volume 4 Issue 1 - 2018 Background information: Medications are the main cause of poisoning in Costa Rica, and benzodiazepines are reported in many of the poisonings that occurred. María del Mar Castillo Guerrero, Freddy Arias Mora Objective: Analyze the poisonings and deaths caused by benzodiazepines in Costa Rica. Department of Pharmacology, University of Costa Rica, Costa Rica Method: A retrospective study was conducted. It included all the cases of poisonings and deaths by benzodiazepine poisonings in Costa Rica from 2007 to 2014. A logistic regression Correspondence: María del Mar Castillo Guerrero, Faculty of analysis was conducted to determine the probability of a person of dying by benzodiazepine Pharmacy, University of Costa Rica, San Pedro, Montes de Oca, poisoning. A descriptive analysis was performed with the information of the total population San José, Costa Rica, Tel 50625118317, Fax 50625118350, of people intoxicated by benzodiazepines. Email [email protected] Results: People intoxicated by benzodiazepines have higher possibilities of dying, Received: February 08, 2018 | Published: February 19, 2018 than those who were not poisoned by benzodiazepines. Most cases of poisoning with benzodiazepines correspond to women in the 30 to 44 age range. Attempting suicide is the main cause of poisoning. The combination of benzodiazepines with other substances such as antidepressants, anticonvulsants and alcohol prevails in cases of deaths by poisoning with benzodiazepines. Discussion and Conclusion: The main cause of poisoning is suicide attempt, so the potential high suicide risk that exists with the use of these drugs should be monitored.