Lessons from Porphyria
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
ANNNNNNNNNNNNNNNNNNNN 100A 006 Left Eye Input Right Eye Input
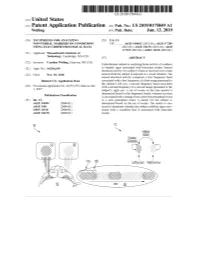
US 20190175049A1 ( 19) United States (12 ) Patent Application Publication (10 ) Pub. No. : US 2019 /0175049 A1 Welling ( 43 ) Pub . Date : Jun . 13 , 2019 ( 54 ) TECHNIQUES FOR ANALYZING (52 ) U . S . CI. NON -VERBAL MARKERS OF CONDITIONS CPC . .. A61B 5 /04842 (2013 . 01 ) ; A61B 5 / 7289 USING ELECTROPHYSIOLOGICAL DATA (2013 . 01) ; A61B 5 /0478 ( 2013 .01 ) ; A61B 5 /7225 ( 2013. 01 ) ; G06N 20 / 10 (2019 .01 ) (71 ) Applicant: Massachusetts Institute of Technology , Cambridge , MA (US ) ( 57 ) ABSTRACT (72 ) Inventor : Caroline Welling, Hanover, NH (US ) Embodiments related to analyzing brain activity of a subject to identify signs associated with binocular rivalry . Sensed ( 21 ) Appl. No. : 16 / 206, 639 electrical activity of a subject' s brain is received over a time period while the subject is exposed to a visual stimulus. The ( 22 ) Filed : Nov. 30 , 2018 sensed electrical activity comprises a first frequency band Related U . S . Application Data associated with a first frequency of a first image presented to the subject ' s left eye , a second frequency band associated (60 ) Provisional application No .62 / 593 , 535, filed on Dec . with a second frequency of a second image presented to the 1 , 2017 subject ' s right eye . A set of events in the time period is determined based on the frequency bands, wherein an event Publication Classification is associated with a change from a previous perceptual event (51 ) Int. Ci. to a new perceptual event. A metric for the subject is A61B 5 /0484 ( 2006 .01 ) determined based on the set of events . The metric is ana A61B 5 /00 ( 2006 .01 ) lyzed to determine whether the subject exhibits signs asso GO6N 20 / 10 (2006 .01 ) ciated with a condition that is associated with binocular A61B 5 /0478 ( 2006 .01 ) rivalry . -

Sulphonmethane, Sulphonal, Diethylsulpho
is a combination of amylene hydrate and chloral hydrate, superior to the corresponding compounds of other elements. while chloralose, a combination of chloral hydrate and glucose, Experience has shown that, in the main, these claims were un- partakes of the action of morphin and is rather expensive. founded, though many, even now, claim that strontium bro- Chloretone, a more recent product, is not entirely devoid of mid disturbs the stomach less than the corresponding sodium danger and is not always so certain in its action as chloral or potassium salt. Another claim that is frequently made by hydrate, while butyl chloral hydrate, or crotón chloral hydrate, manufacturers of nostrums, like "Peacock's Bromides," is that is one of the older compounds that has been found wanting and they use "chemically pure" salts. Exactly what is meant by is now little used. Of the official compounds of this group we this claim is difficult to say, but the Pharmacopeia gives us a have: number of readily applied tests by which the salts themselves Chloralamid and Paraldehyd. may be tested. The manufacturers of nostrums, on the other Chlobalformamidum.—TJ. S.—Chloralformamid. Chlorala- hand, not infrequently add the very substances that are consid- mid. This has practically the same action as therapeutic ered contaminations. doses of chloral hydrate, the latter being formed in the body by {To be continued.) decomposition of chloralformamid. Average dose: 1 gm. (15 grains). Paraldehtdum.—TJ. S.—Paraldehyd is slower in its action transparent liquid, slower in its action than chloral hydrate, but also safer. It has the disadvantage of a persistently dis- A NEW NEEDLE HOLDER. -

Hyperbilirubinemia
Porphyrins Porphyrins (Porphins) are cyclic tetrapyrol compounds formed by the linkage )). of four pyrrole rings through methenyl bridges (( HC In the reduced porphyrins (Porphyrinogens) the linkage of four pyrrole rings (tetrapyrol) through methylene bridges (( CH2 )) The characteristic property of porphyrins is the formation of complexes with the metal ion bound to nitrogen atoms of the pyrrole rings. e.g. Heme (iron porphyrin). Proteins which contain heme ((hemoproteins)) are widely distributed e.g. Hemoglobin, Myoglobin, Cytochromes, Catalase & Tryptophan pyrrolase. Natural porphyrins have substituent side chains on the eight hydrogen atoms numbered on the pyrrole rings. These side chains are: CH 1-Methyl-group (M)… (( 3 )) 2-Acetate-group (A)… (( CH2COOH )) 3-Propionate-group (P)… (( CH2CH2COOH )) 4-Vinyl-group (V)… (( CH CH2 )) Porphyrins with asymmetric arrangement of the side chains are classified as type III porphyrins while those with symmetric arrangement of the side chains are classified as type I porphyrins. Only types I & III are present in nature & type III series is more important because it includes heme. 1 Heme Biosynthesis Heme biosynthesis occurs through the following steps: 1-The starting reaction is the condensation between succinyl-CoA ((derived from citric acid cycle in the mitochondria)) & glycine, this reaction is a rate limiting reaction in the hepatic heme synthesis, it occurs in the mitochondria & is catalyzed by ALA synthase (Aminolevulinate synthase) enzyme in the presence of pyridoxal phosphate as a cofactor. The product of this reaction is α-amino-β-ketoadipate which is rapidly decarboxylated to form δ-aminolevulinate (ALA). 2-In the cytoplasm condensation reaction between two molecules of ALA is catalyzed by ALA dehydratase enzyme to form two molecules of water & one 2 molecule of porphobilinogen (PBG) which is a precursor of pyrrole. -

The Role of Clpx in Erythropoietic Protoporphyria
hematol transfus cell ther. 2018;40(2):182–188 Hematology, Transfusion and Cell Therapy www.rbhh.org Review article The role of ClpX in erythropoietic protoporphyria Jared C. Whitman, Barry H. Paw a, Jacky Chung ∗ Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, United States article info abstract Article history: Hemoglobin is an essential biological component of human physiology and its production Received 28 February 2018 in red blood cells relies upon proper biosynthesis of heme and globin protein. Disruption in Accepted 2 March 2018 the synthesis of these precursors accounts for a number of human blood disorders found Available online 28 March 2018 in patients. Mutations in genes encoding heme biosynthesis enzymes are associated with a broad class of metabolic disorders called porphyrias. In particular, one subtype – erythro- Keywords: poietic protoporphyria – is caused by the accumulation of protoporphyrin IX. Erythropoietic Heme biosynthesis enzymes protoporphyria patients suffer from photosensitivity and a higher risk of liver failure, which Porphyria is the principle cause of morbidity and mortality. Approximately 90% of these patients carry Erythropoietic protoporphyria loss-of-function mutations in the enzyme ferrochelatase (FECH), while 5% of cases are asso- ClpXP ciated with activating mutations in the C-terminus of ALAS2. Recent work has begun to ALAS gene uncover novel mechanisms of heme regulation that may account for the remaining 5% of cases with previously unknown genetic basis. One erythropoietic protoporphyria family has been identified with inherited mutations in the AAA+ protease ClpXP that regulates ALAS activity. In this review article, recent findings on the role of ClpXP as both an activating unfoldase and degrading protease and its impact on heme synthesis will be discussed. -

Aminolevulinic Acid (ALA) As a Prodrug in Photodynamic Therapy of Cancer
Molecules 2011, 16, 4140-4164; doi:10.3390/molecules16054140 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Aminolevulinic Acid (ALA) as a Prodrug in Photodynamic Therapy of Cancer Małgorzata Wachowska 1, Angelika Muchowicz 1, Małgorzata Firczuk 1, Magdalena Gabrysiak 1, Magdalena Winiarska 1, Małgorzata Wańczyk 1, Kamil Bojarczuk 1 and Jakub Golab 1,2,* 1 Department of Immunology, Centre of Biostructure Research, Medical University of Warsaw, Banacha 1A F Building, 02-097 Warsaw, Poland 2 Department III, Institute of Physical Chemistry, Polish Academy of Sciences, 01-224 Warsaw, Poland * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel. +48-22-5992199; Fax: +48-22-5992194. Received: 3 February 2011 / Accepted: 3 May 2011 / Published: 19 May 2011 Abstract: Aminolevulinic acid (ALA) is an endogenous metabolite normally formed in the mitochondria from succinyl-CoA and glycine. Conjugation of eight ALA molecules yields protoporphyrin IX (PpIX) and finally leads to formation of heme. Conversion of PpIX to its downstream substrates requires the activity of a rate-limiting enzyme ferrochelatase. When ALA is administered externally the abundantly produced PpIX cannot be quickly converted to its final product - heme by ferrochelatase and therefore accumulates within cells. Since PpIX is a potent photosensitizer this metabolic pathway can be exploited in photodynamic therapy (PDT). This is an already approved therapeutic strategy making ALA one of the most successful prodrugs used in cancer treatment. Key words: 5-aminolevulinic acid; photodynamic therapy; cancer; laser; singlet oxygen 1. Introduction Photodynamic therapy (PDT) is a minimally invasive therapeutic modality used in the management of various cancerous and pre-malignant diseases. -

International Classification of Diseases
INTERNATIONAL CLASSIFICATION OF DISEASES MANUAL OF THE INTERNATIONAL STATISTICAL CLASSIFICATION OF DISEASES, INJURIES, AND CAUSES OF DEATH Based on the Recommendations of the Eighth Revision Conference, 1965, and Adopted by the Nineteenth World Health Assembly Volume 2 ALPHABETICAL INDEX WORLD HEALTH ORGANIZATION GENEVA 1969 Volume 1 Introduction List of Three-digit Categories Tabular List of Inclusions and Four-digit Sub- categories Medical Certification and Rules for Classification Special Lists for Tabulation Definitions and Recommendations Regulations Volume 2 Alphabetical Index PRINTED IN ENGLAND CONTENTS Introduction Page General arrangement of the Index ....................................... VIII Main sections ............................................................... VIII Structure ..................................................................... IX Code numbzrs .............................................................. x Primary and secondary conditions. ................................... x Multiple diagnoses. ........................................................ XI Spelling....................................................................... XI Order of listing ............................................................. Conventions used in the Index ........................................... XII Parentheses. ................................................................. XII Cross-referexes ........................................................... XI1 Abbreviation NEC. ...................................................... -

REPORT C-Terminal Deletions in the ALAS2 Gene Lead to Gain of Function and Cause X-Linked Dominant Protoporphyria Without Anemia Or Iron Overload
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector REPORT C-Terminal Deletions in the ALAS2 Gene Lead to Gain of Function and Cause X-linked Dominant Protoporphyria without Anemia or Iron Overload Sharon D. Whatley,1,9 Sarah Ducamp,2,3,9 Laurent Gouya,2,3 Bernard Grandchamp,3,4 Carole Beaumont,3 Michael N. Badminton,1 George H. Elder,1 S. Alexander Holme,5 Alexander V. Anstey,5 Michelle Parker,6 Anne V. Corrigall,6 Peter N. Meissner,6 Richard J. Hift,6 Joanne T. Marsden,7 Yun Ma,8 Giorgina Mieli-Vergani,8 Jean-Charles Deybach,2,3,* and Herve´ Puy2,3 All reported mutations in ALAS2, which encodes the rate-regulating enzyme of erythroid heme biosynthesis, cause X-linked sideroblastic anemia. We describe eight families with ALAS2 deletions, either c.1706-1709 delAGTG (p.E569GfsX24) or c.1699-1700 delAT (p.M567EfsX2), resulting in frameshifts that lead to replacement or deletion of the 19–20 C-terminal residues of the enzyme. Prokaryotic expression studies show that both mutations markedly increase ALAS2 activity. These gain-of-function mutations cause a previously unrecognized form of porphyria, X-linked dominant protoporphyria, characterized biochemically by a high proportion of zinc-proto- porphyrin in erythrocytes, in which a mismatch between protoporphyrin production and the heme requirement of differentiating erythroid cells leads to overproduction of protoporphyrin in amounts sufficient to cause photosensitivity and liver disease. Each of the seven inherited porphyrias results from a partial mutations in about 7% of EPP families, of which about deficiency of an enzyme of heme biosynthesis. -

Protecting Mental Health in the Age of Anxiety: the Context of Valium's Development, Synthesis, and Discovery in the United States, to 1963 Catherine (Cai) E
Iowa State University Capstones, Theses and Graduate Theses and Dissertations Dissertations 2009 Protecting mental health in the Age of Anxiety: The context of Valium's development, synthesis, and discovery in the United States, to 1963 Catherine (cai) E. Guise-richardson Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/etd Part of the History Commons Recommended Citation Guise-richardson, Catherine (cai) E., "Protecting mental health in the Age of Anxiety: The onc text of Valium's development, synthesis, and discovery in the United States, to 1963" (2009). Graduate Theses and Dissertations. 10574. https://lib.dr.iastate.edu/etd/10574 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Protecting mental health in the Age of Anxiety: The context of Valium’s development, synthesis, and discovery in the United States, to 1963 by Catherine (Cai) Guise-Richardson A dissertation submitted to the graduate faculty in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Major: History of Technology and Science Program of Study Committee: Hamilton Cravens, Co-Major Professor Alan I Marcus, Co-Major Professor Amy Sue Bix Charles Dobbs David Wilson Iowa State University Ames, Iowa 2009 Copyright © Catherine (Cai) Guise-Richardson, 2009. All rights reserved. ii TABLE OF CONTENTS LIST OF TABLES iv ABSTRACT v CHAPTER 1. -

Modern Surgery, 4Th Edition, by John Chalmers Da Costa Rare Medical Books
Thomas Jefferson University Jefferson Digital Commons Modern Surgery, 4th edition, by John Chalmers Da Costa Rare Medical Books 1903 Modern Surgery - Chapter 23. Diseases and Injuries of the Head John Chalmers Da Costa Jefferson Medical College Follow this and additional works at: https://jdc.jefferson.edu/dacosta_modernsurgery Part of the History of Science, Technology, and Medicine Commons Let us know how access to this document benefits ouy Recommended Citation Da Costa, John Chalmers, "Modern Surgery - Chapter 23. Diseases and Injuries of the Head" (1903). Modern Surgery, 4th edition, by John Chalmers Da Costa. Paper 29. https://jdc.jefferson.edu/dacosta_modernsurgery/29 This Article is brought to you for free and open access by the Jefferson Digital Commons. The Jefferson Digital Commons is a service of Thomas Jefferson University's Center for Teaching and Learning (CTL). The Commons is a showcase for Jefferson books and journals, peer-reviewed scholarly publications, unique historical collections from the University archives, and teaching tools. The Jefferson Digital Commons allows researchers and interested readers anywhere in the world to learn about and keep up to date with Jefferson scholarship. This article has been accepted for inclusion in Modern Surgery, 4th edition, by John Chalmers Da Costa by an authorized administrator of the Jefferson Digital Commons. For more information, please contact: [email protected]. Diseases of the Head 595 XXIII. DISEASES AND INJURIES OF THE HEAD. I. DISEASES OF I:: HEAD. IN approaching a case of brain disorder, first endeavor to locate the seat of the trouble; next, ascertain the nature of the lesion; and, finally, deter- mine the best plan of treatment, operative or otherwise. -

The Many Faces of Apomorphine: Lessons from the Past and Challenges for the Future
Drugs R D (2018) 18:91–107 https://doi.org/10.1007/s40268-018-0230-3 REVIEW ARTICLE The Many Faces of Apomorphine: Lessons from the Past and Challenges for the Future 1 1,2 1,2 Manon Auffret • Sophie Drapier • Marc Ve´rin Published online: 15 March 2018 Ó The Author(s) 2018. This article is an open access publication Abstract Apomorphine is now recognized as the oldest antiparkinsonian drug on the market. Though still under- Key Points used, it is increasingly prescribed in Europe for patients with advanced Parkinson’s disease (PD) with motor fluc- Apomorphine has a long and tortuous path in the tuations. However, its history is far from being limited to therapeutic armamentarium, with numerous movement disorders. This paper traces the history of apo- indications in human and veterinary medicine. morphine, from its earliest empirical use, to its synthesis, The controversy that apomorphine aroused among pharmacological development, and numerous indications clinicians in the past (and in some ways, continues in human and veterinary medicine, in light of its most among neurologists) can be explained by the lack of recent uses and newest challenges. From shamanic rituals controlled studies and its affiliation to morphine. in ancient Egypt and Mesoamerica, to the treatment of There are three main indications for apomorphine: erectile dysfunction, from being discarded as a pharma- emetic, sedative, and antiparkinsonian. cological tool to becoming an essential antiparkinsonian drug, the path of apomorphine in the therapeutic arma- This old drug needs to be reconsidered by clinicians mentarium has been tortuous and punctuated by setbacks and will benefit from current galenic and technical and groundbreaking discoveries. -

A High Urinary Urobilinogen / Serum Total Bilirubin Ratio Reported in Abdominal Pain Patients Can Indicate Acute Hepatic Porphyria
A High Urinary Urobilinogen / Serum Total Bilirubin Ratio Reported in Abdominal Pain Patients Can Indicate Acute Hepatic Porphyria Chengyuan Song Shandong University Qilu Hospital Shaowei Sang Shandong University Qilu Hospital Yuan Liu ( [email protected] ) Shandong University Qilu Hospital https://orcid.org/0000-0003-4991-552X Research Keywords: acute hepatic porphyria, urinary urobilinogen, serum total bilirubin Posted Date: June 14th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-587707/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/10 Abstract Background: Due to its variable symptoms and nonspecic laboratory test results during routine examinations, acute hepatic porphyria (AHP) has always been a diagnostic dilemma for physicians. Misdiagnoses, missed diagnoses, and inappropriate treatments are very common. Correct diagnosis mainly depends on the detection of a high urinary porphobilinogen (PBG) level, which is not a routine test performed in the clinic and highly relies on the physician’s awareness of AHP. Therefore, identifying a more convenient indicator for use during routine examinations is required to improve the diagnosis of AHP. Results: In the present study, we retrospectively analyzed laboratory examinations in 12 AHP patients and 100 patients with abdominal pain of other causes as the control groups between 2015 and 2021. Compared with the control groups, AHP patients showed a signicantly higher urinary urobilinogen level during the urinalysis (P < 0.05). However, we showed that the higher urobilinogen level was caused by a false- positive result due to a higher level of urine PBG in the AHP patients. Hence, we used serum total bilirubin, an upstream substance of urinary urobilinogen synthesis, for calibration. -

A Mouse Model of Hereditary Coproporphyria Identified in an ENU Mutagenesis Screen Ashlee J
© 2017. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2017) 10, 1005-1013 doi:10.1242/dmm.029116 RESEARCH ARTICLE A mouse model of hereditary coproporphyria identified in an ENU mutagenesis screen Ashlee J. Conway1, Fiona C. Brown1, Robert O. Fullinfaw2, Benjamin T. Kile3, Stephen M. Jane1,4 and David J. Curtis1,* ABSTRACT (Bissell and Wang, 2015). Clinically, porphyria usually emerges in A genome-wide ethyl-N-nitrosourea (ENU) mutagenesis screen in adolescence with acute attacks triggered by factors that activate mice was performed to identify novel regulators of erythropoiesis. hepatic enzymes, such as fasting, alcohol, sulphonamide antibiotics, Here, we describe a mouse line, RBC16, which harbours a and hormones such as progesterone (Bissell and Wang, 2015). dominantly inherited mutation in the Cpox gene, responsible for Biochemically, HCP presents with a marked increase in porphyrin production of the haem biosynthesis enzyme, coproporphyrinogen III precursors, such as porphobilinogen (PBG), as well as porphyrins, oxidase (CPOX). A premature stop codon in place of a tryptophan at notably coproporphyrinogen III, which accumulate and are detected amino acid 373 results in reduced mRNA expression and diminished in urine and faeces in high concentrations during episodic attacks, but protein levels, yielding a microcytic red blood cell phenotype in can be normal or only marginally elevated during latent periods. heterozygous mice. Urinary and faecal porphyrins in female RBC16 Avoidance of known triggers is so far the only approach to managing heterozygotes were significantly elevated compared with that of wild- the acute hepatic porphyrias. Symptoms can be alleviated with type littermates, particularly coproporphyrinogen III, whereas males substances that inhibit haem biosynthesis, such as glucose loading were biochemically normal.