Safety and Effectiveness of the UV-X System for Corneal Collagen Cross
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

LCD): Computerized Corneal Topography (L33810
Local Coverage Determination (LCD): Computerized Corneal Topography (L33810) Links in PDF documents are not guaranteed to work. To follow a web link, please use the MCD Website. Contractor Information CONTRACTOR NAME CONTRACT TYPE CONTRACT NUMBER JURISDICTION STATE(S) First Coast Service Options, Inc. A and B MAC 09102 - MAC B J - N Florida First Coast Service Options, Inc. A and B MAC 09202 - MAC B J - N Puerto Rico First Coast Service Options, Inc. A and B MAC 09302 - MAC B J - N Virgin Islands LCD Information Document Information LCD ID Original Effective Date L33810 For services performed on or after 10/01/2015 LCD Title Revision Effective Date Computerized Corneal Topography For services performed on or after 01/08/2019 Proposed LCD in Comment Period Revision Ending Date N/A N/A Source Proposed LCD Retirement Date N/A N/A AMA CPT / ADA CDT / AHA NUBC Copyright Notice Period Start Date Statement N/A CPT codes, descriptions and other data only are copyright 2019 American Medical Association. All Rights Notice Period End Date Reserved. Applicable FARS/HHSARS apply. N/A Current Dental Terminology © 2019 American Dental Association. All rights reserved. Copyright © 2019, the American Hospital Association, Chicago, Illinois. Reproduced with permission. No portion of the AHA copyrighted materials contained within this publication may be copied without the express written consent of the AHA. AHA copyrighted materials including the UB-04 codes and descriptions may not be removed, copied, or utilized within any Created on 01/02/2020. Page 1 of 7 software, product, service, solution or derivative work without the written consent of the AHA. -

Division of Health Care Financing & Policy SB 278 Section 16
Division of Health Care Financing & Policy SB 278 Section 16 - Physician Rates Reporting Facility & Non-Facility Rate Comparison Nevada 2016 2016 Medicaid Medicaid Medicaid Medicare Medicare vs. vs. Procedure Code & Description Rates (1) Non-Facility Facility Medicare Medicare Rates for Rates for Non-Facility Facility 10021 Fna w/o image 70.92 128.90 72.80 (57.98) (1.88) 10022 Fna w/image 65.39 148.21 68.38 (82.82) (2.99) 10030 Guide cathet fluid drainage 154.73 827.01 176.71 (672.28) (21.98) 10035 Perq dev soft tiss 1st imag 86.35 568.38 90.92 (482.03) (4.57) 10036 Perq dev soft tiss add imag 43.52 495.42 45.82 (451.90) (2.30) 10040 Acne surgery 87.47 106.03 92.10 (18.56) (4.63) 10060 Drainage of skin abscess 95.60 122.68 101.60 (27.08) (6.00) 10061 Drainage of skin abscess 178.04 215.50 188.01 (37.46) (9.97) 10080 Drainage of pilonidal cyst 103.29 188.83 107.87 (85.54) (4.58) 10081 Drainage of pilonidal cyst 172.45 281.57 177.64 (109.12) (5.19) 10120 Remove foreign body 103.22 159.56 108.73 (56.34) (5.51) 10121 Remove foreign body 185.58 287.08 194.44 (101.50) (8.86) 10140 Drainage of hematoma/fluid 118.06 170.87 124.18 (52.81) (6.12) 10160 Puncture drainage of lesion 95.62 136.49 100.72 (40.87) (5.10) 10180 Complex drainage wound 178.97 258.94 188.14 (79.97) (9.17) 11000 Debride infected skin 28.42 56.88 29.76 (28.46) (1.34) 11001 Debride infected skin add-on 14.22 22.40 14.87 (8.18) (0.65) 11004 Debride genitalia & perineum 579.35 608.28 608.28 (28.93) (28.93) 11005 Debride abdom wall 781.65 822.97 822.97 (41.32) (41.32) 11006 Debride -

Current Developments in Corneal Topography and Tomography
diagnostics Review Current Developments in Corneal Topography and Tomography Piotr Kanclerz 1,2,* , Ramin Khoramnia 3 and Xiaogang Wang 4 1 Hygeia Clinic, Department of Ophthalmologyul, Ja´skowaDolina 57, 80-286 Gda´nsk,Poland 2 Helsinki Retina Research Group, University of Helsinki, 00100 Helsinki, Finland 3 The David J. Apple International Laboratory for Ocular Pathology, Department of Ophthalmology, University of Heidelberg, 69120 Heidelberg, Germany; [email protected] 4 Department of Cataract, Shanxi Eye Hospital, Taiyuan 030002, China; [email protected] * Correspondence: [email protected] Abstract: Introduction: Accurate assessment of the corneal shape is important in cataract and refractive surgery, both in screening of candidates as well as for analyzing postoperative outcomes. Although corneal topography and tomography are widely used, it is common that these technologies are confused. The aim of this study was to present the current developments of these technologies and particularly distinguish between corneal topography and tomography. Methods: The PubMed, Web of Science and Embase databases were the main resources used to investigate the medical literature. The following keywords were used in various combinations: cornea, corneal, topography, tomography, Scheimpflug, Pentacam, optical coherence tomography. Results: Topography is the study of the shape of the corneal surface, while tomography allows a three-dimensional section of the cornea to be presented. Corneal topographers can be divided into large- and small-cone Placido-based devices, as well as devices with color-LEDs. For corneal tomography, scanning slit or Scheimpflug imaging and optical coherence tomography may be employed. In several devices, corneal topography and tomography have been successfully combined with tear-film analysis, aberrometry, optical biometry and anterior/posterior segment optical coherence tomography. -

Refractive Surgery
I explore and get information from inside the eye, that´s my job. You can then go deeper into the diagnosis. You decide, I explore! FOR ACCURACY IN REFRACTIVE SURGERY ACE® is a technology that utilizes the power of high-resolution swept-source OCT imaging to provide the key corneal measurements. Optimizing the quality of the preoperative data provides more information to help you to improve the safety of your refractive surgery procedures.1 ACE® and the TECHNOLAS® TeneoTM 317 Model 2 offer solutions that will refine your results. Transform your daily surgical routine into an exciting day with a platform that brings together corneal topography and tomography and allowing data transfer between both devices. All corneal measurements are based on high-resolution swept-source OCT images KEY FUNCTIONS Corneal topography Corneal wavefront analysis Corneal tomography Differential maps Pachymetry Progression analysis Total corneal power Data transfer with TECHNOLAS® TENEOTM 317 Model 2 * All corneal measurements based on high- resolution swept-source OCT images 1. Muriël Doors et al. Value of optical coherence tomography for anterior segment surgery. J Cataract Refract Surg 2010; 36:1213–1229 Q 2010 ADVANCED CORNEAL EXPLORER HIGHLY CUSTOMIZABLE MAP LAYOUT Display up to 6 maps simultaneously, compare OD and OS, or perform an analysis over time. 12 different map types: Assess each patient’s corneal topography and Anterior and posterior axial or Pachymetry tomography, including tangential curvature Total corneal power curvature and elevation Anterior and posterior elevation maps of the anterior and Anterior and total corneal wavefront (best fit sphere and best fit torus) posterior surfaces. -
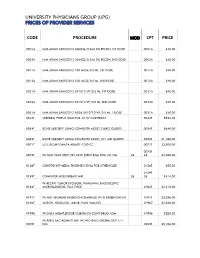
Code Procedure Cpt Price University Physicians Group
UNIVERSITY PHYSICIANS GROUP (UPG) PRICES OF PROVIDER SERVICES CODE PROCEDURE MOD CPT PRICE 0001A IMM ADMN SARSCOV2 30MCG/0.3ML DIL RECON 1ST DOSE 0001A $40.00 0002A IMM ADMN SARSCOV2 30MCG/0.3ML DIL RECON 2ND DOSE 0002A $40.00 0011A IMM ADMN SARSCOV2 100 MCG/0.5 ML 1ST DOSE 0011A $40.00 0012A IMM ADMN SARSCOV2 100 MCG/0.5 ML 2ND DOSE 0012A $40.00 0021A IMM ADMN SARSCOV2 5X1010 VP/0.5 ML 1ST DOSE 0021A $40.00 0022A IMM ADMN SARSCOV2 5X1010 VP/0.5 ML 2ND DOSE 0022A $40.00 0031A IMM ADMN SARSCOV2 AD26 5X10^10 VP/0.5 ML 1 DOSE 0031A $40.00 0042T CEREBRAL PERFUS ANALYSIS, CT W/CONTRAST 0042T $954.00 0054T BONE SURGERY USING COMPUTER ASSIST, FLURO GUIDED 0054T $640.00 0055T BONE SURGERY USING COMPUTER ASSIST, CT/ MRI GUIDED 0055T $1,188.00 0071T U/S LEIOMYOMATA ABLATE <200 CC 0071T $2,500.00 0075T 0075T PR TCAT PLMT XTRC VRT CRTD STENT RS&I PRQ 1ST VSL 26 26 $2,208.00 0126T CAROTID INT-MEDIA THICKNESS EVAL FOR ATHERSCLER 0126T $55.00 0159T 0159T COMPUTER AIDED BREAST MRI 26 26 $314.00 PR RECTAL TUMOR EXCISION, TRANSANAL ENDOSCOPIC 0184T MICROSURGICAL, FULL THICK 0184T $2,315.00 0191T PR ANT SEGMENT INSERTION DRAINAGE W/O RESERVOIR INT 0191T $2,396.00 01967 ANESTH, NEURAXIAL LABOR, PLAN VAG DEL 01967 $2,500.00 01996 PR DAILY MGMT,EPIDUR/SUBARACH CONT DRUG ADM 01996 $285.00 PR PERQ SAC AGMNTJ UNI W/WO BALO/MCHNL DEV 1/> 0200T NDL 0200T $5,106.00 PR PERQ SAC AGMNTJ BI W/WO BALO/MCHNL DEV 2/> 0201T NDLS 0201T $9,446.00 PR INJECT PLATELET RICH PLASMA W/IMG 0232T HARVEST/PREPARATOIN 0232T $1,509.00 0234T PR TRANSLUMINAL PERIPHERAL ATHERECTOMY, RENAL -

Comparison of Corneal Topography in Eyes Before and One Month After the Phacoemulsification Procedure
Research Article ISSN: 2574 -1241 DOI: 10.26717/BJSTR.2020.25.004275 Comparison of Corneal Topography in Eyes Before and One Month After the Phacoemulsification Procedure Pateras Evangelos1* and Armenis J2 1Associate Professor, Biomedical Sciences Department, Course Optics & Optometry, University of West Attica, Greece 2Armenis J, Ophthalmologist, MSC candidate, Biomedical Sciences Departmentt, Course Optics & Optometry, University of West Attica, Greece *Corresponding author: Pateras Evangelos, Associate Professor, O O, Biomedical Sciences Department, Course Optics & Optometry, University of West Attica, Greece ARTICLE INFO Abstract Received: February 13, 2020 Antares Placido topographer CSO had been used to measure 50 eyes corneas at Published: February 24, 2020 Ophthalmological Clinic of the Greek Red Cross hospital «Korgialeneio - Benakeio in collaboration with University of West Attica and their refractive maps were taken. The refractive power map was taken before cataract surgery with the method of phaco- Citation: Pateras Evangelos, Armenis J. emulsification. The keratometric measurements for 3 & 5mm were recorded before and Comparison of Corneal Topography in one month after surgery. The correlation of the K1, K2 readings before and one month after the phacoemulsification procedure, do not cause changes in the refractive power of Eyes Before and One Month After the the two main meridians of the cornea of patients, in order to alter the final astigmatism refractive post-operative effect, to a statistically significant degree. J Sci & Tech Res 25(5)-2020. BJSTR. Phacoemulsification Procedure. Biomed Keywords: Cornea; Main Meridians; Astigmatism; With the Rule; Against the Rule; MS.ID.004275. Surgical Incisions; Phacoemulsification; Topography; Correlation Purpose ranging from 2 to 28 µm [11-13]. -

Evaluation of Corneal Shape and Biomechanics Before LASIK
Evaluation of Corneal Shape and Biomechanics Before LASIK Renato Ambro´sio, Jr, MD, PhD Leonardo P. Nogueira, MD Diogo L. Caldas, MD Bruno M. Fontes, MD Allan Luz, MD Jorge O. Cazal, MD Milton Ruiz Alves, MD, PhD Michael W. Belin, MD, FACS ’ Introduction The preoperative evaluation is of critical importance for success in laser in situ keratomileusis (LASIK). This examination should fulfill 3 main purposes: counseling and educating the candidates, surgery planning, and screening for cases at higher risk for complications. It is critical to interview each refractive patient to assess their individual needs and to provide realistic expectations. A thorough ophthalmologic examination is mandatory, including specific complementary examina- tions to characterize many aspects of the cornea and the optics of the eye. In fact, it is notable that refractive surgery has motivated tremendous development for advanced diagnostic methods, among many others advancements and innovations in Ophthalmology.1,2 One of the most important aspects of the preoperative examination of LASIK candidates is to screen cases at risk for progressive ectasia. INTERNATIONAL OPHTHALMOLOGY CLINICS Volume 51, Number 2, 11–39 r 2011, Lippincott Williams & Wilkins www.internat-ophthalmology.com | 11 12 ’ Ambro´ sio et al Keratectasia has emerged as a rare but very severe complication of LASIK, which is a leading cause of litigation.3,4 In post-LASIK ectasia, the lamellar cut and excimer laser ablation lead to a state of bio- mechanical failure with an inability to support the continuous stresses caused by intraocular pressure (IOP), extraocular muscle action, blinking, eye rubbing, and other forces.5 The corneal stroma undergoes a 2-step process of delamination and interfibril fracture, which is clinically characterized by thinning and bulging of the cornea. -

Irregular Astigmatism After Corneal Transplantation—Efficacy And
CLINICAL SCIENCE Irregular Astigmatism After Corneal Transplantation—Efficacy and Safety of Topography-Guided Treatment Inês Laíns, MD, MSc,*† Andreia M. Rosa, MD,*† Marta Guerra, MD, MSc,* Cristina Tavares, MD,* Conceição Lobo, MD, PhD,*† Maria F. L. Silva, PhD,‡ Maria J. Quadrado, MD, PhD,*† and Joaquim N. Murta, MD, PhD*† Key Words: astigmatism, corneal topography, corneal transplanta- Purpose: fi To analyze the ef cacy and safety of topography-guided tion, excimer, photorefractive keratectomy photorefractive keratectomy (TG-PRK) to treat irregular astigmatism after corneal transplantation. (Cornea 2016;35:30–36) Methods: This was a retrospective observational case series. Eyes with irregular astigmatism after penetrating keratoplasty treated with orneal transplantation (CT) is one of the most commonly TG-PRK (Allegretto Wave Eye-Q) with the topography-guided Cperformed transplant procedures throughout the world.1 customized ablation treatment protocol were included. All treatments Although it is generally successful, patients frequently present had been planned to correct the topographic irregularities, as well as with high irregular postoperative astigmatism, which pre- to reduce the refractive error after neutralizing the induced refractive cludes them from achieving satisfactory visual acuity.2 It can change. Clinical records, treatment plan, and the examinations affect up to 40% of the eyes3 and remains a challenge even for performed were reviewed and the following data were collected: experienced cornea specialists, as spectacles and rigid contact corrected and uncorrected distance visual acuities; manifest refrac- lenses often represent an unsuccessful option for visual tion; topographic parameters, and corneal endothelial cell count. rehabilitation.4 Results: We included 31 eyes [30 patients; mean age 45.0 6 13.4 In recent decades, excimer laser treatment has emerged 5 (SD) years]. -

Keratoconus and Corneal Stability After Radial Keratectomy in the Fellow Eye
Relato de Caso | CASE REPORT Keratoconus and corneal stability after radial keratectomy in the fellow eye: case report Ceratocone e estabilidade corneana após ceratectomia radial no outro olho: relato de caso JACQUELINE MARTINS SOUSA1, FLAVIO EDUARDO HIRAI1, ELCIO HIDEO SATO1 ABSTRACT RESUMO Keratoconus has usually been described as bilateral but asymmetric disease. Cor- O ceratocone é descrito como uma doença bilateral porém assimétrica e vários dados neal ectasia is one of the long-term complications of modern refractive surgery, na literatura comprovam que a ectasia corneana é uma das complicações de longo especially those submitted to laser in situ keratomileusis (LASIK). We describe a prazo da cirurgia refrativa moderna, especialmente do laser in situ keratomileusis patient with keratoconus in the right eye that was submitted to radial keratectomy (LASIK). Nós descrevemos um caso de uma paciente com ceratocone no olho direito (RK) in the left eye 19 years ago with no progression of the ectatic cornea and no e que foi submetida à ceratotomia radial no olho esquerdo há 19 anos, desde então complications related to the refractive surgery. Because unilateral keratoconus is sem sinais de progressão da ectasia corneana nem de complicações relativas à cirurgia rare, we believe that RK was performed on an already ectatic cornea (not clinically refrativa. Como o ceratocone unilateral é raro, acreditamos que a cirurgia refrativa detected) or with fruste keratoconus. However, neither corneal ectasia progressed, tenha sido realizada num olho com ectasia corneana não detectada clinicamente ou nor ectasia was induced by RK in the fellow eye. com ceratocone frustro. Entretanto, a ectasia do olho direito não progrediu e também não houve sinais de ectasia no olho submetido à cirurgia refrativa nesse período de 19 anos de acompanhamento. -

Topography-Guided Laser Assisted
White Paper Topography-Guided Laser Assisted In-Situ Keratomileusis vs Small-Incision Lenticule Extraction Refractive Surgery — A Summary of Clinical Outcomes Andrea Petznick, Diplom-AO (FH), PhD1 Victor Higuera, OD2 1. Alcon Medical Affairs, North America 2. Alcon Global Training Manager for Surgical Lasers Introduction The global procedure volume for refractive surgery was approximately 4.3 million in 2018. Of those procedures, 774,000 were performed in the US market.1 Modern refractive surgery has evolved significantly since the era of Radial Keratotomy, and with a projected growth of up to 5.5 million procedures globally in 20231, it is important to provide a clinical perspective of the latest available refractive surgery modalities. Key Take-aways: • Alcon WaveLight® Topography-Guided platform outperforms small-incision lenticule extraction (SMILE*) • When compared to LASIK, SMILE has not conclusively shown that the procedure has greater biomechanical stability and results in reduced post-operative dry eye symptoms as a short-term consequence of higher nerve fiber preservation • Alcon WaveLight® Topography-Guided platform improves quality of vision and results in a high patient satisfaction Clinical challenge: Maintaining the natural shape of the cornea Refractive Surgery reduces refractive error by reshaping the corneal surface. The ultimate goal of refractive surgery is to eliminate patient’s dependency on glasses or contact lenses. Since refractive surgery is an elective surgery, there are many options for the surgeon to recommend and the patient to choose from, including but not limited to, Photorefractive Keratectomy (PRK), Laser Assisted In-situ Keratomileusis (LASIK) or Small Incision Lenticule Extraction (SMILE) to name a few. -

IPA Par Ophthalmology Reimbursement
IPA Par Ophthalmology Reimbursement CMO reimburses its IPA par Ophthalmologists at a maximum per visit rate of $212.00, for services billed during a single comprehensive eye exam. A Comprehensive Eye exam is considered as including an initial or subsequent eye exam, including refraction and exam under anesthesia; initial or established Evaluation and Management (E&M) exam; Tonography; Visual Field tests and Ophthalmoscopy. A Comprehensive Eye Exam will include the following individually billed services: Procedure Description Code(s) 92002 – 92004 Initial Eye Exam 92012 – 92020 Subsequent, including Refraction, 92015 and exam under anesthesia 92081 - 92083 Visual Field tests 92100 Serial Tonometry (separate procedure) 92201 Ophthalmoscopy, extended; with retinal drawing and scleral depression of peripheral retinal disease (eg, for retinal tear, retinal detachment, retinal tumor) with interpretation and report, unilateral or bilateral 92202 Ophthalmoscopy, extended; with drawing of optic nerve or macula (eg, for glaucoma, macular pathology, tumor) with interpretation and report, unilateral or bilateral 92225 - 92226 Ophthalmoscopy 99202 – 99205 Initial E&M (Evaluation and Management) 99211 – 99215 Established E&M 99241 – 99245 Consultations – NOT Covered. Bill with Initial or Established E&M or Initial/Subsequent Eye Exam codes. Vision testing services not included in the above per visit fee maximum will be reimbursed at the fee for service level and pay 80% of the prevailing Bronx-based Medicare fee schedule, in addition to the per visit -

Refractive Lens Surgery Mark Packer, MDT, I
Ophthalmol Clin N Am 19 (2006) 77 – 88 Refractive Lens Surgery Mark Packer, MDT, I. Howard Fine, MD, Richard S. Hoffman, MD Drs. Fine, Hoffman & Packer, 1550 Oak Street, Suite 5, Eugene OR 97401, USA Axial length measurement remains an indispens- IOLMaster (Zeiss Humphrey Systems, Jena, Ger- able technique for intraocular lens (IOL) power many) with measurements obtained by immersion calculation. Recently, partial coherence interferome- ultrasound using the Axis II (Quantel Medical, try has emerged as a new modality for biometry [1]. Clermont-Ferrand, France). The postoperative refrac- The postoperative results achieved with this mo- tions of patients undergoing cataract extraction with dality have been considered analogous to those posterior chamber IOL implantation were also exam- achieved with the ultrasound immersion technique [2]. ined to determine the accuracy of the immersion Although reportedly ‘‘user-friendly’’ and less depen- ultrasound technique. dent on technician expertise than are ultrasound Fifty cataractous eyes underwent preoperative methods, noncontact optical biometry is limited by axial length measurement with both the Axis II and dense media such as posterior subcapsular cataract. A IOLMaster. For the Axis II immersion technique, the second limitation of the optical method is the lack of Praeger shell was employed. Patients were placed in a a lens thickness measurement, which is a required sitting position in an examination room chair with the variable in the Holladay II IOL power calculation head reclined gently against the headrest. The average software, version 2.30.9705. According to Holladay, ‘‘Total Length’’ reported by the unit was entered into the lens thickness can be estimated by the formula, the Holladay II IOL power calculation formula.