Micro RNA and Non-Coding DNA and Repeats
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

National Institutional Ranking Framework
National Institutional Ranking Framework Ministry of Human Resource Development Government of India Welcome to Data Capturing System: OVERALL Submitted Institute Data for NIRF'2020' Institute Name: ACADEMY OF SCIENTIFIC & INNOVATIVE RESEARCH [IR-O-U-0713] Sanctioned (Approved) Intake Academic Year 2018-19 2017-18 2016-17 2015-16 2014-15 2013-14 PG [1 Year Program(s)] 17 - - - - - PG [2 Year Program(s)] 136 65 - - - - Total Actual Student Strength (Program(s) Offered by Your Institution) (All programs No. of Male No. of Female Total Students Within State Outside State Outside Economically Socially No. of students No. of students No. of students No. of students of all years) Students Students (Including male (Including male Country Backward Challenged receiving full receiving full receiving full who are not & female) & female) (Including male (Including male (SC+ST+OBC tuition fee tuition fee tuition fee receiving full & female) & female) Including male reimbursement reimbursement reimbursement tuition fee & female) from the State from Institution from the Private reimbursement and Central Funds Bodies Government PG [1 Year 6 3 9 2 7 0 0 2 0 0 0 2 Program(s)] PG [2 Year 53 56 109 48 61 0 1 42 1 1 0 41 Program(s)] Placement & Higher Studies PG [1 Years Program(s)]: Placement & higher studies for previous 3 years Academic Year No. of first year No. of first year Academic Year No. of students graduating in minimum No. of students Median salary of No. of students students intake in the students admitted in stipulated time placed placed selected for Higher year the year graduates(Amount in Studies Rs.) 2016-17 20 20 2016-17 17 5 319000(Rupees Three 9 Lakhs Nineteen Thousand Only) 2017-18 16 16 2017-18 16 10 330000(Rupees Three 2 Lakhs Thirty Thousand Only) 2018-19 17 9 2018-19 9 2 325000(Rupees Three 4 Lakhs Twenty Five Thousand Only) PG [2 Years Program(s)]: Placement & higher studies for previous 3 years Academic Year No. -

Office of the Public Relations Officer- Media Coordinator Jamia Millia Islamia
Office of the Public Relations Officer- Media Coordinator Jamia Millia Islamia December 26, 2018 Press Release Jamia Professor honored with National Bioscience Award by Department of Biotechnology, Govt of India Prof. Mohammad Zahid Ashraf has been honoured with National Bio-science Award 2018 as recognition of his research excellence. This award instituted by the Department of Biotechnology of the Government of India recognizes research and development work carried out in India during the last 5 years of the career. The award is given to Indian bio-scientists of less than 45 years of age, who has made unique contributions towards the development of state of art in basic and applied areas of biological sciences. The award carries a citation, a plaque, a cash prize of Rs 300,000 and a research support grant of Rs 15,00,000 for three years. It is considered as one of the highest Indian science awards and as equivalent to the Shanti Swarup Bhatnagar Prize given by the Council of Scientific and Industrial Research of India. Dr. Ashraf is currently a Professor in the Department of Biotechnology at Faculty of Natural Sciences, Jamia Millia Islamia (JMI), New Delhi. Prior to this he was Head of the Genomics Division at Defence Institute of Physiology and Allied Sciences (DIPAS), Defence Research and Development Organization (DRDO), Delhi. Dr. Ashraf is pioneer in the field of high altitude thrombosis and has done novel work in resolving the mystery of blood clotting on exposure to hypoxia at high altitudes in Soldiers posted at extreme altitudes including Siachen Glacier. His research has unraveled many problems associated with high altitude thrombosis and mechanisms associated with its pathology. -
Annual Report 2017-18
The National Academy of Sciences, India (NASI) Annual Report (April 2017 – March 2018) 5, Lajpatrai Road, Allahabad - 211 002 i POSTAL ADDRESS The National Academy of Sciences, India 5, Lajpatrai Road, Allahabad – 211002, India PHONE +91-532-2640224, 2441243 FAX +91-532-2641183 E-MAIL [email protected] WEBSITE http://www.nasi.nic.in http://www.nasi.org.in NASI, Allahabad is also on the FACEBOOK & TWITTER Published by the General Secretary (HQ), NASI for The National Academy of Sciences, India, Allahabad ii Late Prof. Meghnad Saha, Founder President An Academy of Science can do a great deal by educating public opinion, undertaking particular problems, and bringing out scientific workers in various fields for discussion and cooperative research. But the main function of the Academy should be towards cultural improvement by contributions to human knowledge. - Prof. Meghnad Saha on the Inaugural Session of the Academy India is justified in feeling proud for its unique contributions to science in ancient days. However, successive foreign invasions and alien rule for centuries pushed science in the background and the country went through with what may be described as the dark age for science. Western science attracted Indian intelligentsia after the establishment of the western system of education and the universities; and despite many constraints, the country could produce giants like Prof. Meghnad Saha, Prof. S. N. Bose, Sir J.C. Bose and Acharya Prafulla Chandra Ray. The first World War and the world-wide economic depression caused a set back to scientific research globally - much more so in India whose scientists found it difficult even to publish their research work since they had to be almost entirely dependent on foreign journals. -

9-March 2021.Xlsx
VENKATESHWAR INTERNATIONAL SCHOOL Sector -10, Dwarka, New Delhi - 110075 LIST OF CANDIDATES WHO APPLIED FOR ADMISSION UNDER OPEN SEATS (2021-2022) FOR PRE SCHOOL/NURSERY S.No. RegNo Student Name Date of Birth Gender Father Name Mother Name 1 2021/0001 Vanya Talwar 06.08.2017 Female Arjun Talwar Garima Talwar 2 2021/0002 Jaiteg Singh Anand 04.07.2017 Male Sarabjit Singh Anand Jasmeet Kaur Anand 3 2021/0003 Nibhiv Chadha 08.03.2018 Male Dr Nishant Chadha Priyanka Chadha 4 2021/0004 Taksh Bajaj 26.07.2017 Male Manish Bajaj Rekha Mirchandani 5 2021/0005 Mahira Singh 08.09.2017 Female Dr Ram Singh Dr Anita 6 2021/0006 Yakshit Anand 27.09.2017 Male Roushan Anand Neha Kumari 7 2021/0007 Madhav Sharma 07.04.2017 Male Amit Sharma Isha Kaushik 8 2021/0008 Hridaan Nagar 06.04.2017 Male Sumit Nagar Rupali Nagar 9 2021/0009 Namish Mishra 09.11.2017 Male Vikram Mishra Alka Mishra 10 2021/0010 Ojas Batra 08.08.2017 Male Yogesh Batra Liza Rajpal 11 2021/0011 Dakshita Khattar 29.11.2017 Female Pankaj Khattar Palak Khaneja 12 2021/0012 Akse Yusuf Khan 29.10.2017 Female Md Shahnawaz Khan Shabishtan Zaman Khanam 13 2021/0013 Arayna Verma 19.08.2017 Female Vishal Verma Pallak Verma 14 2021/0014 Ojasvi Upadhyay 09.10.2017 Female Aditya Upadhyaya Anupam Priyadarshini 15 2021/0015 Manaksh Chauhan 25.08.2017 Male Manish Chauhan Akhilesh Rajpoot 16 2021/0016 Aviram Singh 05.11.2017 Male Sambhu Singh Anu Singh 17 2021/0017 Disha Jain 17.03.2018 Female Anshul Jain Sonia Kansal 18 2021/0018 Samarth Kumar 09.03.2018 Male Prashant Kumar Lovely Singh 19 2021/0019 Samriddhi -

Curriculum Vitae
Curriculum Vitae Prof. SAMIR K. BRAHMACHARI Director General, Council of Scientific and Industrial Research. Anusandhan Bhawan, 2, Rafi Marg, New Delhi-10001,INDIA Phone: 91-11-2371 0472, 2371 7053 Fax : 91-11-2371 0618 Email : [email protected]; [email protected] Born: January, 1, 1952; Nationality: Indian Current position Director General, Council of Scientific and Industrial Research, New Delhi, Scientist and Founder Director, CSIR- Institute of Genomics and Integrative Biology, Delhi, Chief Mentor, Open Source Drug Discovery Unit, CSIR, New Delhi. Academics 1972 Bachelor of Science in Chemistry (Hons), University of Calcutta. 1974 Master of Science in Chemistry (Specialisation, Physical Chemistry), University of Calcutta. 1978 PhD, Molecular Biophysics Unit, Indian Institute of Science, Bangalore. 1978-1979 Post Doctoral Research, French Govt. Fellowship, Institute de Recherche en Biologie Moleculaire, CNRS, University of Paris VIII. 1979-1981 Research Associate, Molecular Biophysics Unit, Indian Institute of Science, Bangalore. 1981-1997 Lecturer/ Assistant/Associate Professor, Molecular Biophysics Unit and Associated Faculty, Centre for Genetic Engineering, Indian Institute of Science, Bangalore. 1997-2001 Professor, Molecular Biophysics Unit, Indian Institute of Science, Bangalore. 1997-2007 Director, CSIR-Institute of Genomics and Integrative Biology, Delhi. 2003 Adjunct Professor, Bioinformatics Centre, University of Pune, Pune. 2007 Director General, Council of Scientific and Industrial Research (CSIR), New Delhi. 2013 Academy Professor, Academy of Scientific and Industrial Research (AcSIR), New Delhi Visiting Scientist Nov 1979 Dept of Biochemistry, Memorial University of Newfoundland, Canada. June-Sept Department of Genetics and Development, Columbia University New York, USA. 1985 Visiting Professorship May-June 1997 Max Planck Institute of Biophysical Chemistry, Goettingen, Germany. -

PROF. SAMIR K. BRAHMACHARI Date of Birth
Last updated 16 September 2011 CURRICULUM VITAE Name : PROF. SAMIR K. BRAHMACHARI Date of Birth : 1st January 1952 Present Position : Secretary, Department of Scientific and Industrial Research, GOI & Director General, Council of Scientific and Industrial Research. Anusandhan Bhawan, 2, Rafi Ahmed Kidwai Marg, New Delhi-110001 INDIA Phone : 91-11-2371 0472, 2371 7053 Fax : 91-11-2371 0618 E-mail : [email protected]; [email protected] Academic Record Degree Subject Year University Ph.D. Molecular Biophysics 1978 Indian Institute of Science, Bangalore M.Sc. Pure Chemistry 1973-74 University of Calcutta (Physical Chemistry Specialisation) B.Sc. Chemistry (Hons.) 1971-72 University of Calcutta i .exe Professional Appointments Positions Held Period Name of Organization Secretary, DSIR & November 2007- Present Secretary, Department of Director General, Scientific and CSIR Industrial Research, Govt. of India. & Director General, Council of Scientific and Industrial Research Director 11th August 1997-November Institute of Genomics & 2007 Integrative Biology (CSIR), Delhi Professor June 1997 - August 2001 Molecular Biophysics Unit, Indian Institute of Science, Bangalore Associate April 1992-June1997 Molecular Biophysics Unit, Professor Indian Institute of Science, Bangalore Visiting Professor May-July 1997 Max. Planck Institute of Biophysical Chemistry, Goettingen, Germany Associated April 1989-March 1995 Centre for Genetic Engineering, Faculty Indian Institute of Science, Bangalore. Asst. Professor April 1986 - March 1992 Molecular Biophysics Unit, Indian Institute of Science, Bangalore Lecturer April 1981 - March 1986 Molecular Biophysics Unit, Indian Institute of Science, Bangalore Research December 1979-March 1981 Molecular Biophysics Unit, Associate Indian Institute of Science, Bangalore Visiting Scientist November 1979 Dept of Biochemistry, Memorial University of Newfoundland, St. -

Retrovirology Biomed Central
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Springer - Publisher Connector Retrovirology BioMed Central Review Open Access Host-virus interaction: a new role for microRNAs Vinod Scaria, Manoj Hariharan, Souvik Maiti, Beena Pillai and Samir K Brahmachari* Address: GN Ramachandran Knowledge Center for Genome Informatics, Institute of Genomics and Integrative Biology, CSIR, Mall Road, Delhi 110 007, India Email: Vinod Scaria - [email protected]; Manoj Hariharan - [email protected]; Souvik Maiti - [email protected]; Beena Pillai - [email protected]; Samir K Brahmachari* - [email protected] * Corresponding author Published: 11 October 2006 Received: 30 August 2006 Accepted: 11 October 2006 Retrovirology 2006, 3:68 doi:10.1186/1742-4690-3-68 This article is available from: http://www.retrovirology.com/content/3/1/68 © 2006 Scaria et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract MicroRNAs (miRNAs) are a new class of 18–23 nucleotide long non-coding RNAs that play critical roles in a wide spectrum of biological processes. Recent reports also throw light into the role of microRNAs as critical effectors in the intricate host-pathogen interaction networks. Evidence suggests that both virus and hosts encode microRNAs. The exclusive dependence of viruses on the host cellular machinery for their propagation and survival also make them highly susceptible to the vagaries of the cellular environment like small RNA mediated interference. -

NIRF 2021 Rankings Data (Under “Research Institutions
National Institutional Ranking Framework Ministry of Education Government of India Welcome to Data Capturing System: OVERALL Submitted Institute Data for NIRF'2021' Institute Name: ACADEMY OF SCIENTIFIC & INNOVATIVE RESEARCH [IR-O-U-0713] Sanctioned (Approved) Intake Academic Year 2019-20 2018-19 2017-18 2016-17 2015-16 2014-15 PG [1 Year Program(s)] 60 - - - - - PG [2 Year Program(s)] 160 136 - - - - Total Actual Student Strength (Program(s) Offered by Your Institution) (All programs No. of Male No. of Female Total Students Within State Outside State Outside Economically Socially No. of students No. of students No. of students No. of students of all years) Students Students (Including male (Including male Country Backward Challenged receiving full receiving full receiving full who are not & female) & female) (Including male (Including male (SC+ST+OBC tuition fee tuition fee tuition fee receiving full & female) & female) Including male reimbursement reimbursement reimbursement tuition fee & female) from the State from Institution from the Private reimbursement and Central Funds Bodies Government PG [1 Year 9 4 13 4 9 0 1 6 0 0 0 7 Program(s)] PG [2 Year 75 80 155 85 70 0 7 60 0 0 0 67 Program(s)] Placement & Higher Studies PG [1 Years Program(s)]: Placement & higher studies for previous 3 years Academic Year No. of first year No. of first year Academic Year No. of students graduating in minimum No. of students Median salary of No. of students students intake in the students admitted in stipulated time placed placed selected for Higher year the year graduates(Amount in Studies Rs.) 2017-18 16 16 2017-18 16 10 330000(Rupees Three 2 Lakhs Thirty Thousand Only) 2018-19 17 9 2018-19 9 2 325000(Rupees Three 4 Lakhs Twenty Five Thousand Only) 2019-20 60 13 2019-20 10 8 375000(Rupees Three 0 Lakhs Seventy Five Thousand Only) PG [2 Years Program(s)]: Placement & higher studies for previous 3 years Academic Year No. -

Last Updated 10 December 2010
Last updated 10 December 2010 Curriculum Vitae Name : Professor. SAMIR K. BRAHMACHARI Date of Birth : 1st January 1952 Present Position : Secretary, Department of Scientific and Industrial Research, GOI & Director General, Council of Scientific and Industrial Research. Anusandhan Bhawan, 2, Rafi Ahmed Kidwai Marg, New Delhi-110001 INDIA Phone : 91-11-2371 0472, 2371 7053 Fax : 91-11-2371 0618 E-mail : [email protected]; [email protected] Academic Record Degree Subject Year University Ph.D. Molecular Biophysics 1978 Indian Institute of Science, Bangalore M.Sc. Pure Chemistry 1973-74 University of Calcutta (Physical Chemistry Specialisation) B.Sc. Chemistry (Hons.) 1971-72 University of Calcutta Prof. Samir K. Brahmachari Page 1 of 47 i.exe Professional Appointments Positions Held Period Name of Organization Secretary, DSIR & November 2007- Present Secretary, Department of Scientific and Director General, Industrial Research, Govt. of India. & CSIR Director General, Council of Scientific and Industrial Research Director 11th August 1997-November 2007 Institute of Genomics & Integrative Biology (CSIR), Delhi Professor June 1997 – August 2001 Molecular Biophysics Unit, Indian Institute of Science, Bangalore Associate Professor April 1992-June1997 Molecular Biophysics Unit, Indian Institute of Science, Bangalore Visiting Professor May-July 1997 Max. Planck Institute of Biophysical Chemistry, Goettingen, Germany Associated Faculty April 1989-March 1995 Centre for Genetic Engineering, Indian Institute of Science, Bangalore. Asst. Professor April 1986 - March 1992 Molecular Biophysics Unit, Indian Institute of Science, Bangalore Lecturer April 1981 - March 1986 Molecular Biophysics Unit, Indian Institute of Science, Bangalore Research Associate December 1979-March 1981 Molecular Biophysics Unit, Indian Institute of Science, Bangalore Visiting Scientist November 1979 Dept of Biochemistry, Memorial University of Newfoundland, St. -

VENKATESHWAR INTERNATIONAL SCHOOL Sector
VENKATESHWAR INTERNATIONAL SCHOOL Sector - 10, Dwarka, New Delhi - 110075 POINT-WISE LIST OF APPLICANTS FOR PRE-SCHOOL ADMISSION (2021-2022) NOTE: 1) There will be a draw for applicants with 70 points on 17th March, 2021 at 3.00 pm in the school grounds. Please carry your registration slip for the draw. 2) Parent's are requested to follow all appropriate Covid behaviour. Wearing of masks is mandatory. Please maintain social distancing and carry a bottle of sanitizer with you. Single parent representative is requested to attend to avoid overcrowding. 3) Applicants with 90 and 80 points are selected for admission to Preschool. 4) In case of duplicate entries, the latest registration number has been considered. Total S.NO. Student Name DateofBirth Gender Mother Name Father Name Reg No Distance Alumini Sibling Points 1 Dhanisha Arora 22.11.2017 Female Nidhi Arora Harish Arora 2021/0045 70 0 20 90 2 Taraansh Mittal 17.11.2017 Male Ankita Mittal Nitin Mittal 2021/0081 70 0 20 90 3 Zohaan Ullah Chauhan 26.09.2017 Male Amreen Khan Talat Ullah Chauhan 2021/0106 70 0 20 90 4 Kaashvi Sharma 01.11.2017 Female Aditi Mishra Keshav Sharma 2021/0145 70 0 20 90 5 Aadhya Jain 25.08.2017 Female Ritu Jain Nitin Jain 2021/0168 70 0 20 90 6 Shivika Singhal 08.03.2018 Female Neha Singhal Ajay Singhal 2021/0258 70 0 20 90 7 Rik Joshi 22.08.2017 Male Preeti Joshi Atri Joshi 2021/0261 70 0 20 90 8 Inaaya Sikka 28.04.2017 Female Swati Sikka Gaurav Sikka 2021/0281 70 0 20 90 9 Maira Khan 02.01.2018 Female Priyanki Khan Nadeem Khan 2021/0342 70 0 20 90 10 Aarav Gogia -

Acknowledgements
ACKNOWLEDGEMENTS I am completing my sixth year at IGIB when I am writing this acknowledgement. This journey of my PhD where I have given most productive 6 years of my life has taught me a lot of things and transformed me as a person. Most important lessons I have learnt from here is, a never give up attitude, and how to rise after repeated failure thanks to crystallography. I have started my PhD in crystallography area and ending it in a genomics area. This would not have been possible in any other institute in India. So I am greatly indebted to my research institute IGIB and all the people who supported me during my journey of PhD. Although words might not be enough to express my gratitude towards all the people who helped me during this journey but I will still try. First, I would like to give my sincere regards to Dr. Rakesh Sharma, for registering me as a PhD student under his guidance and for his expert scientific advices. I would like to acknowledge most important person for my training and PhD my mentor Dr. Bhupesh Taneja, it would not have been possible for me to finish my work without his constant support, guidance and motivation. His professional skills and strong background in structural biology helped me defining and completing my PhD objectives. Most important lessons I learnt from him apart from science is how to “keep calm and carry on” in adverse situation. I really appreciate the way he gave me scientific thinking space outside his core expertise, and his eagerness and willingness to support me for learning novel techniques through collaborations. -
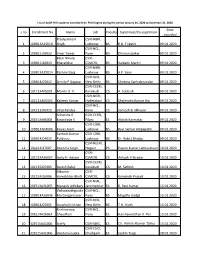
S.No Enrollment No. Name Lab Faculty Supervisor/Co-Supervisor Date Awarded 1 10BB13A25010 Pradyumna K Singh CSIR-NBRI, Lucknow B
List of AcSIR PhD students awarded their PhD Degree during the period January 01, 2020 to December 31, 2020 Date s.no Enrollment No. Name Lab Faculty Supervisor/Co-supervisor awarded Pradyumna K CSIR-NBRI, 1 10BB13A25010 Singh Lucknow BS R.D. Tripathi 09.01.2020 CSIR-NCL, 2 10BB15J26032 Amar Yewar Pune BS Dhiman Sarkar 09.01.2020 Balar Nikunj CSIR- 3 10BB15J16013 Bharatbhai CSMCRI, BS Vaibahv Mantri 09.01.2020 CSIR-NBRI, 4 10BB13A25014 Rashmi Garg Lucknow BS A.P. Sane 09.01.2020 CSIR-IGIB, 5 10BB14J02012 Urmila P Gagtap New Delhi BS Chetana Sachidanandan 09.01.2020 CSIR-CECRI, 6 10CC14A05001 Manila O. V. Karaikudi CS A. Subbiah 09.01.2020 CSIR-NGRI, 7 10CC14A05005 Kaleesh Kumar Hyderabad CS Shailendra Kumar Jha 09.01.2020 CSIR-NCL, 8 10CC13J26020 Virat Pandya Pune CS Santosh B. Mhaske 09.01.2020 Sriharsha R CSIR-CEERI, 9 20EE13A06003 Koundinya V Pilani ES Abhijit Karmakar 09.01.2020 CSIR-CDRI, 10 10BB13A04006 Faiyaz Alam Lucknow BS Ravi Sankar Ampapathi 09.01.2020 Santosh Kumar CSIR-CDRI, 11 10BB14J04011 Puttrevu Lucknow BS Dr. Rabi S Bhatta 09.01.2020 CSIR-NEERI, 12 10pp13J27007 Akansha Singh Nagpur PS Pawan Kumar Labhasetwar 10.01.2020 CSIR- 13 10CC14A16007 Jacky H. Advani CSMCRI, CS Ankush V Biradar 10.01.2020 CSIR-CECRI, 14 10CC15J05005 Suresh Balaji Karaikudi CS M. Sathish 10.01.2020 Jitkumar CSIR- 15 10CC14J16006 Hareshbhai Bhatt CSMCRI, CS Kamalesh Prasad 10.01.2020 CSIR-NML, 16 20EE13A31007 Manashi Adhikary Jamshedpur ES B. Ravi Kumar 15.01.2020 Vishwanathgouda CSIR-NCL, 17 10BB14A26046 Maralingannavar Pune BS Mugdha Gadgil 15.01.2020 CSIR-IGIB, 18 10BB14J02001 Anusha Krishnan New Delhi BS T.N.