Building and Mining an Atlas of the Mammalian Circadian Transcriptome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Minimal 16Q Genomic Loss Implicates Cadherin-11 in Retinoblastoma
Minimal 16q Genomic Loss Implicates Cadherin-11 in Retinoblastoma Mellone N. Marchong,1,2 Danian Chen,1,6 Timothy W. Corson,1,3 Cheong Lee,1 Maria Harmandayan,1 Ella Bowles,1 Ning Chen,5 and Brenda L. Gallie1,2,3,4,5 1Divisions of Cancer Informatics and Cellular and Molecular Biology, Ontario Cancer Institute/Princess Margaret Hospital, University Health Network, Toronto, Ontario, Canada; Departments of 2Medical Biophysics, 3Molecular and Medical Genetics, and 4Ophthalmology, University of Toronto, Toronto, Ontario, Canada; 5Retinoblastoma Solutions, Toronto, Ontario, Canada; and 6Department of Ophthalmology, West China Hospital, Faculty of Medicine, Sichuan University, Chengdu, People’s Republic of China Abstract compared with normal adult human retina. Our analyses Retinoblastoma is initiated by loss of both RB1 alleles. implicate CDH11, but not CDH13, as a potential tumor Previous studies have shown that retinoblastoma suppressor gene in retinoblastoma. (Mol Cancer Res tumors also show further genomic gains and losses. We 2004;2(9):495–503) now define a 2.62 Mbp minimal region of genomic loss of chromosome 16q22, which is likely to contain tumor suppressor gene(s), in 76 retinoblastoma tumors, using Introduction loss of heterozygosity (30 of 76 tumors) and quantitative Retinoblastoma is the most common intraocular tumor in multiplex PCR (71 of 76 tumors). The sequence-tagged children. Mutations of both alleles (M1 and M2) of the RB1 site WI-5835 within intron 2 of the cadherin-11 (CDH11) gene at chromosome 13q14 are necessary (1) for retinoblastoma gene showed the highest frequency of loss (54%, 22 of tumor initiation but not sufficient for malignant transformation 41 samples tested). -

Chromosomal Microarray Analysis in Turkish Patients with Unexplained Developmental Delay and Intellectual Developmental Disorders
177 Arch Neuropsychitry 2020;57:177−191 RESEARCH ARTICLE https://doi.org/10.29399/npa.24890 Chromosomal Microarray Analysis in Turkish Patients with Unexplained Developmental Delay and Intellectual Developmental Disorders Hakan GÜRKAN1 , Emine İkbal ATLI1 , Engin ATLI1 , Leyla BOZATLI2 , Mengühan ARAZ ALTAY2 , Sinem YALÇINTEPE1 , Yasemin ÖZEN1 , Damla EKER1 , Çisem AKURUT1 , Selma DEMİR1 , Işık GÖRKER2 1Faculty of Medicine, Department of Medical Genetics, Edirne, Trakya University, Edirne, Turkey 2Faculty of Medicine, Department of Child and Adolescent Psychiatry, Trakya University, Edirne, Turkey ABSTRACT Introduction: Aneuploids, copy number variations (CNVs), and single in 39 (39/123=31.7%) patients. Twelve CNV variant of unknown nucleotide variants in specific genes are the main genetic causes of significance (VUS) (9.75%) patients and 7 CNV benign (5.69%) patients developmental delay (DD) and intellectual disability disorder (IDD). were reported. In 6 patients, one or more pathogenic CNVs were These genetic changes can be detected using chromosome analysis, determined. Therefore, the diagnostic efficiency of CMA was found to chromosomal microarray (CMA), and next-generation DNA sequencing be 31.7% (39/123). techniques. Therefore; In this study, we aimed to investigate the Conclusion: Today, genetic analysis is still not part of the routine in the importance of CMA in determining the genomic etiology of unexplained evaluation of IDD patients who present to psychiatry clinics. A genetic DD and IDD in 123 patients. diagnosis from CMA can eliminate genetic question marks and thus Method: For 123 patients, chromosome analysis, DNA fragment analysis alter the clinical management of patients. Approximately one-third and microarray were performed. Conventional G-band karyotype of the positive CMA findings are clinically intervenable. -

Genome-Wide Association Study of Growth Performance and Immune Response to Newcastle Disease Virus of Indigenous Chicken in Rwanda
ORIGINAL RESEARCH published: 16 August 2021 doi: 10.3389/fgene.2021.723980 Genome-Wide Association Study of Growth Performance and Immune Response to Newcastle Disease Virus of Indigenous Chicken in Rwanda Richard Habimana 1,2*, Kiplangat Ngeno 2, Tobias Otieno Okeno 2, Claire D’ andre Hirwa 3, Christian Keambou Tiambo 4 and Nasser Kouadio Yao 5 1 College of Agriculture, Animal Science and Veterinary Medicine, University of Rwanda, Kigali, Rwanda, 2 Animal Breeding and Genomics Group, Department of Animal Science, Egerton University, Egerton, Kenya, 3 Rwanda Agricultural and Animal Resources Development Board, Kigali, Rwanda, 4 Centre for Tropical Livestock Genetics and Health, International Livestock Research Institute, Nairobi, Kenya, 5 Biosciences Eastern and Central Africa – International Livestock Research Institute (BecA-ILRI) Hub, Nairobi, Kenya A chicken genome has several regions with quantitative trait loci (QTLs). However, Edited by: replication and confirmation of QTL effects are required particularly in African chicken Younes Miar, Dalhousie University, populations. This study identified single nucleotide polymorphisms (SNPs) and putative Canada genes responsible for body weight (BW) and antibody response (AbR) to Newcastle Reviewed by: disease (ND) in Rwanda indigenous chicken (IC) using genome-wide association studies Sayed Haidar Abbas, Northwest A & F University, China (GWAS). Multiple testing was corrected using chromosomal false detection rates of 5 and Suxu Tan, 10% for significant and suggestive thresholds, respectively. BioMart data mining and Michigan State University, variant effect predictor tools were used to annotate SNPs and candidate genes, respectively. United States A total of four significant SNPs (rs74098018, rs13792572, rs314702374, and rs14123335) *Correspondence: Richard Habimana significantly (p ≤ 7.6E−5) associated with BW were identified on chromosomes (CHRs) [email protected] 8, 11, and 19. -

Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations Fall 2010 Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress Renuka Nayak University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Computational Biology Commons, and the Genomics Commons Recommended Citation Nayak, Renuka, "Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress" (2010). Publicly Accessible Penn Dissertations. 1559. https://repository.upenn.edu/edissertations/1559 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/1559 For more information, please contact [email protected]. Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress Abstract Genes interact in networks to orchestrate cellular processes. Here, we used coexpression networks based on natural variation in gene expression to study the functions and interactions of human genes. We asked how these networks change in response to stress. First, we studied human coexpression networks at baseline. We constructed networks by identifying correlations in expression levels of 8.9 million gene pairs in immortalized B cells from 295 individuals comprising three independent samples. The resulting networks allowed us to infer interactions between biological processes. We used the network to predict the functions of poorly-characterized human genes, and provided some experimental support. Examining genes implicated in disease, we found that IFIH1, a diabetes susceptibility gene, interacts with YES1, which affects glucose transport. Genes predisposing to the same diseases are clustered non-randomly in the network, suggesting that the network may be used to identify candidate genes that influence disease susceptibility. -

Genome-Wide Association Analysis in Humans Links Nucleotide Metabolism to Leukocyte Telomere Length
http://www.diva-portal.org This is the published version of a paper published in American Journal of Human Genetics. Citation for the original published paper (version of record): Li, C., Stoma, S., Lotta, L A., Warner, S., Albrecht, E. et al. (2020) Genome-wide Association Analysis in Humans Links Nucleotide Metabolism to Leukocyte Telomere Length American Journal of Human Genetics, 106(3): 389-404 https://doi.org/10.1016/j.ajhg.2020.02.006 Access to the published version may require subscription. N.B. When citing this work, cite the original published paper. Permanent link to this version: http://urn.kb.se/resolve?urn=urn:nbn:se:umu:diva-169391 ARTICLE Genome-wide Association Analysis in Humans Links Nucleotide Metabolism to Leukocyte Telomere Length Chen Li,1,3,85 Svetlana Stoma,2,3,85 Luca A. Lotta,1,85 Sophie Warner,2,85 Eva Albrecht,4 Alessandra Allione,5,6 Pascal P. Arp,7 Linda Broer,7 Jessica L. Buxton,8,9 Alexessander Da Silva Couto Alves,10,11 Joris Deelen,12,13 Iryna O. Fedko,14 Scott D. Gordon,15 Tao Jiang,16 Robert Karlsson,17 Nicola Kerrison,1 Taylor K. Loe,18 Massimo Mangino,19,20 Yuri Milaneschi,21 Benjamin Miraglio,22 Natalia Pervjakova,23 Alessia Russo,5,6 Ida Surakka,22,24 Ashley van der Spek,25 Josine E. Verhoeven,21 Najaf Amin,25 Marian Beekman,13 Alexandra I. Blakemore,26,27 Federico Canzian,28 Stephen E. Hamby,2,3 Jouke-Jan Hottenga,14 Peter D. Jones,2 Pekka Jousilahti,29 Reedik Ma¨gi,23 Sarah E. -

'Dynamic Gene Expression Response to Altered Gravity in Human
www.nature.com/scientificreports OPEN Dynamic gene expression response to altered gravity in human T cells Cora S. Thiel1,2, Swantje Hauschild1,2, Andreas Huge3, Svantje Tauber1,2, Beatrice A. Lauber1, Jennifer Polzer1, Katrin Paulsen1, Hartwin Lier4, Frank Engelmann4,5, Burkhard Schmitz6, 6 1 1,2,7,8 Received: 1 February 2017 Andreas Schütte , Liliana E. Layer & Oliver Ullrich Accepted: 31 May 2017 We investigated the dynamics of immediate and initial gene expression response to different Published: xx xx xxxx gravitational environments in human Jurkat T lymphocytic cells and compared expression profiles to identify potential gravity-regulated genes and adaptation processes. We used the Affymetrix GeneChip® Human Transcriptome Array 2.0 containing 44,699 protein coding genes and 22,829 non-protein coding genes and performed the experiments during a parabolic flight and a suborbital ballistic rocket mission to cross-validate gravity-regulated gene expression through independent research platforms and different sets of control experiments to exclude other factors than alteration of gravity. We found that gene expression in human T cells rapidly responded to altered gravity in the time frame of 20 s and 5 min. The initial response to microgravity involved mostly regulatory RNAs. We identified three gravity-regulated genes which could be cross-validated in both completely independent experiment missions: ATP6V1A/D, a vacuolar H + -ATPase (V-ATPase) responsible for acidification during bone resorption, IGHD3-3/IGHD3-10, diversity genes of the immunoglobulin heavy-chain locus participating in V(D)J recombination, and LINC00837, a long intergenic non-protein coding RNA. Due to the extensive and rapid alteration of gene expression associated with regulatory RNAs, we conclude that human cells are equipped with a robust and efficient adaptation potential when challenged with altered gravitational environments. -

Application of Microarray-Based Comparative Genomic Hybridization in Prenatal and Postnatal Settings: Three Case Reports
SAGE-Hindawi Access to Research Genetics Research International Volume 2011, Article ID 976398, 9 pages doi:10.4061/2011/976398 Case Report Application of Microarray-Based Comparative Genomic Hybridization in Prenatal and Postnatal Settings: Three Case Reports Jing Liu, Francois Bernier, Julie Lauzon, R. Brian Lowry, and Judy Chernos Department of Medical Genetics, University of Calgary, 2888 Shaganappi Trail NW, Calgary, AB, Canada T3B 6A8 Correspondence should be addressed to Judy Chernos, [email protected] Received 16 February 2011; Revised 20 April 2011; Accepted 20 May 2011 Academic Editor: Reha Toydemir Copyright © 2011 Jing Liu et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Microarray-based comparative genomic hybridization (array CGH) is a newly emerged molecular cytogenetic technique for rapid evaluation of the entire genome with sub-megabase resolution. It allows for the comprehensive investigation of thousands and millions of genomic loci at once and therefore enables the efficient detection of DNA copy number variations (a.k.a, cryptic genomic imbalances). The development and the clinical application of array CGH have revolutionized the diagnostic process in patients and has provided a clue to many unidentified or unexplained diseases which are suspected to have a genetic cause. In this paper, we present three clinical cases in both prenatal and postnatal settings. Among all, array CGH played a major discovery role to reveal the cryptic and/or complex nature of chromosome arrangements. By identifying the genetic causes responsible for the clinical observation in patients, array CGH has provided accurate diagnosis and appropriate clinical management in a timely and efficient manner. -

A Genome-Wide Association Study Identifies UTRN Gene Polymorphism for Restless Legs Syndrome in a Korean Population
ORIGINAL ARTICLE Print ISSN 1738-3684 / On-line ISSN 1976-3026 https://doi.org/10.4306/pi.2017.14.6.830 OPEN ACCESS A Genome-Wide Association Study Identifies UTRN Gene Polymorphism for Restless Legs Syndrome in a Korean Population Chul-Hyun Cho1*, Ji-Hye Choi2*, Seung-Gul Kang3, Ho-Kyoung Yoon1, Young-Min Park4, Joung-Ho Moon1, Ki-Young Jung5, Jin-Kyu Han6, Hong-Bum Shin7, Hyun Ji Noh8, Yong Seo Koo9, Leen Kim1, Hyun Goo Woo2 , and Heon-Jeong Lee1 1Department of Psychiatry, Korea University College of Medicine, Seoul, Republic of Korea 2Department of Physiology, Ajou University School of Medicine, Suwon, Republic of Korea 3Department of Psychiatry, School of Medicine, Gachon University, Incheon, Republic of Korea 4Department of Psychiatry, Inje University, Ilsan Paik Hospital, Goyang, Republic of Korea 5Department of Neurology, Seoul University College of Medicine, Seoul, Republic of Korea 6Seoul Sleep Center, Seoul, Republic of Korea 7Kosleep Clinic, Seoul, Republic of Korea 8Broad Institute of MIT and Harvard, Cambridge, MA, USA 9Department of Neurology, Korea University College of Medicine, Seoul, Republic of Korea ObjectiveaaRestless legs syndrome (RLS) is a highly heritable and common neurological sensorimotor disease disturbing sleep. The ob- jective of study was to investigate significant gene for RLS by performing GWA and replication study in a Korean population. MethodsaaWe performed a GWA study for RLS symptom group (n=325) and non-RLS group (n=2,603) from the Korea Genome Epide- miology Study. We subsequently performed a replication study in RLS and normal controls (227 RLS and 229 controls) to confirm the present GWA study findings as well as previous GWA study results. -

Case Report Application of Microarray-Based Comparative Genomic Hybridization in Prenatal and Postnatal Settings: Three Case Reports
SAGE-Hindawi Access to Research Genetics Research International Volume 2011, Article ID 976398, 9 pages doi:10.4061/2011/976398 Case Report Application of Microarray-Based Comparative Genomic Hybridization in Prenatal and Postnatal Settings: Three Case Reports Jing Liu, Francois Bernier, Julie Lauzon, R. Brian Lowry, and Judy Chernos Department of Medical Genetics, University of Calgary, 2888 Shaganappi Trail NW, Calgary, AB, Canada T3B 6A8 Correspondence should be addressed to Judy Chernos, [email protected] Received 16 February 2011; Revised 20 April 2011; Accepted 20 May 2011 Academic Editor: Reha Toydemir Copyright © 2011 Jing Liu et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Microarray-based comparative genomic hybridization (array CGH) is a newly emerged molecular cytogenetic technique for rapid evaluation of the entire genome with sub-megabase resolution. It allows for the comprehensive investigation of thousands and millions of genomic loci at once and therefore enables the efficient detection of DNA copy number variations (a.k.a, cryptic genomic imbalances). The development and the clinical application of array CGH have revolutionized the diagnostic process in patients and has provided a clue to many unidentified or unexplained diseases which are suspected to have a genetic cause. In this paper, we present three clinical cases in both prenatal and postnatal settings. Among all, array CGH played a major discovery role to reveal the cryptic and/or complex nature of chromosome arrangements. By identifying the genetic causes responsible for the clinical observation in patients, array CGH has provided accurate diagnosis and appropriate clinical management in a timely and efficient manner. -

Loci for Human Leukocyte Telomere Length in the Singaporean Chinese Population and Trans-Ethnic Genetic Studies
ARTICLE https://doi.org/10.1038/s41467-019-10443-2 OPEN Loci for human leukocyte telomere length in the Singaporean Chinese population and trans-ethnic genetic studies Rajkumar Dorajoo 1,14, Xuling Chang2,3,14, Resham Lal Gurung4,14, Zheng Li 1, Ling Wang1, Renwei Wang5, Kenneth B. Beckman 6, Jennifer Adams-Haduch5, Yiamunaa M4, Sylvia Liu4, Wee Yang Meah1, Kar Seng Sim 1, Su Chi Lim4,7,8, Yechiel Friedlander9, Jianjun Liu 1,10, Rob M. van Dam8,10, Jian-Min Yuan 5,11,15, Woon-Puay Koh8,12,15, Chiea Chuen Khor 1,13,15 & Chew-Kiat Heng 2,3,15 1234567890():,; Genetic factors underlying leukocyte telomere length (LTL) may provide insights into telo- mere homeostasis, with direct links to disease susceptibility. Genetic evaluation of 23,096 Singaporean Chinese samples identifies 10 genome-wide loci (P <5×10−8). Several of these contain candidate genes (TINF2, PARP1, TERF1, ATM and POT1) with potential roles in telo- mere biology and DNA repair mechanisms. Meta-analyses with additional 37,505 European individuals reveals six more genome-wide loci, including associations at MPHOSPH6, NKX2-3 and TYMS. We demonstrate that longer LTL associates with protection against respiratory disease mortality [HR = 0.854(0.804–0.906), P = 1.88 × 10−7] in the Singaporean Chinese samples. We further show that the LTL reducing SNP rs7253490 associates with respiratory infections (P = 7.44 × 10−4) although this effect may not be strongly mediated through LTL. Our data expands on the genetic basis of LTL and may indicate on a potential role of LTL in immune competence. -

Estimating Telomere Length from Whole Genome Sequencing Data
Estimating telomere length from whole genome sequencing data J. H. R. Farmery CRUK: CI University of Cambridge This dissertation is submitted for the degree of Doctor of Philosophy Christ’s College May 2018 For my Parents who put me on the right track. and for Alexandra who kept me on it Declaration This dissertation is the result of my own work and includes nothing which is the outcome of work done in collaboration except where specifically indicated in the text. It is not substantially the same as any that I have submitted, or, is being concurrently submitted for a degree or diploma or other qualification at the University of Cambridge or any other University or similar institution except as declared in the Preface and specified in the text. I further state that no substantial part of my dissertation has already been submitted, or, is being concurrently submitted for any such degree, diploma or other qualification at the University of Cambridge or any other University of similar institution except as declared in the Preface and specified in the text. This dissertation does not exceed the 60,000 word limit prescribed by the Clinical Medicine and Clinical Veterinary Medicine Degree Committee. J. H. R. Farmery May 2018 Acknowledgements I would first like to acknowledge my parents, Moira and David, as well as my sisters, Olivia and Sophie, for their limitless support and encouragement. Also Libor Juranek and Alexandra Weissova Snr. for their hospitality and kindness throughout my time at Cambridge. Thanks are also due to my friends, in Cambridge and elsewhere, for being such great chums. -
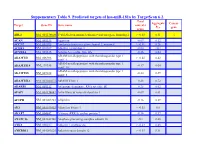
Suppementary Table 9. Predicted Targets of Hsa-Mir-181A by Targetscan 6.2
Suppementary Table 9. Predicted targets of hsa-miR-181a by TargetScan 6.2. Total Aggregate Cancer Target Gene ID Gene name context+ P gene score CT ABL2 NM_001136000 V-abl Abelson murine leukemia viral oncogene homolog 2 > -0.03 0.31 √ ACAN NM_001135 Aggrecan -0.09 0.12 ACCN2 NM_001095 Amiloride-sensitive cation channel 2, neuronal > -0.01 0.26 ACER3 NM_018367 Alkaline ceramidase 3 -0.04 <0.1 ACVR2A NM_001616 Activin A receptor, type IIA -0.16 0.64 ADAM metallopeptidase with thrombospondin type 1 ADAMTS1 NM_006988 > -0.02 0.42 motif, 1 ADAM metallopeptidase with thrombospondin type 1 ADAMTS18 NM_199355 -0.17 0.64 motif, 18 ADAM metallopeptidase with thrombospondin type 1 ADAMTS5 NM_007038 -0.24 0.59 motif, 5 ADAMTSL1 NM_001040272 ADAMTS-like 1 -0.26 0.72 ADARB1 NM_001112 Adenosine deaminase, RNA-specific, B1 -0.28 0.62 AFAP1 NM_001134647 Actin filament associated protein 1 -0.09 0.61 AFTPH NM_001002243 Aftiphilin -0.16 0.49 AK3 NM_001199852 Adenylate kinase 3 > -0.02 0.6 AKAP7 NM_004842 A kinase (PRKA) anchor protein 7 -0.16 0.37 ANAPC16 NM_001242546 Anaphase promoting complex subunit 16 -0.1 0.66 ANK1 NM_000037 Ankyrin 1, erythrocytic > -0.03 0.46 ANKRD12 NM_001083625 Ankyrin repeat domain 12 > -0.03 0.31 ANKRD33B NM_001164440 Ankyrin repeat domain 33B -0.17 0.35 ANKRD43 NM_175873 Ankyrin repeat domain 43 -0.16 0.65 ANKRD44 NM_001195144 Ankyrin repeat domain 44 -0.17 0.49 ANKRD52 NM_173595 Ankyrin repeat domain 52 > -0.05 0.7 AP1S3 NM_001039569 Adaptor-related protein complex 1, sigma 3 subunit -0.26 0.76 Amyloid beta (A4) precursor protein-binding, family A, APBA1 NM_001163 -0.13 0.81 member 1 APLP2 NM_001142276 Amyloid beta (A4) precursor-like protein 2 -0.05 0.55 APOO NM_024122 Apolipoprotein O -0.32 0.41 ARID2 NM_152641 AT rich interactive domain 2 (ARID, RFX-like) -0.07 0.55 √ ARL3 NM_004311 ADP-ribosylation factor-like 3 > -0.03 0.51 ARRDC3 NM_020801 Arrestin domain containing 3 > -0.02 0.47 ATF7 NM_001130059 Activating transcription factor 7 > -0.01 0.26 ATG2B NM_018036 ATG2 autophagy related 2 homolog B (S.