Calibrating the in Vitro–In Vivo Correlation for OATP-Mediated Drug-Drug Interactions with Rosuvastatin Using Static and PBPK Models S
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Preferred Drug List
January 2012 Preferred Drug List The Preferred Drug List, administered by CVS Caremark on behalf of Marriott International, is a guide within select therapeutic categories for clients, plan members and health care providers. Generics should be considered the first line of prescribing. If there is no generic available, there may be more than one brand-name medicine to treat a condition. These preferred brand-name medicines are listed to help identify products that are clinically appropriate and cost-effective. Generics listed in therapeutic categories are for representational purposes only. This is not an all-inclusive list. This list represents brand products in CAPS and generic products in lowercase italics. PLAN MEMBER HEALTH CARE PROVIDER Your benefit plan provides you with a prescription benefit program Your patient is covered under a prescription benefit plan administered by CVS Caremark. Ask your doctor to consider administered by CVS Caremark. As a way to help manage health prescribing, when medically appropriate, a preferred medicine from care costs, authorize generic substitution whenever possible. If you this list. Take this list along when you or a covered family member believe a brand-name product is necessary, consider prescribing a sees a doctor. brand name on this list. Please note: Please note: • Your specific prescription benefit plan design may not cover • Generics should be considered the first line of prescribing. certain categories, regardless of their appearance in this document. • This drug list represents a summary of prescription coverage. It is not inclusive and does not guarantee coverage. • For specific information regarding your prescription benefit coverage and copay1 information, please visit • The member's prescription benefit plan may have a different www.caremark.com or contact a CVS Caremark Customer copay for specific products on the list. -

PRESCRIBING INFORMATION OGEN* (Estropipate) Tablets 0.75 Mg, 1.5
PRESCRIBING INFORMATION OGEN* (estropipate) Tablets 0.75 mg, 1.5 mg, 3.0 mg Estrogen Pfizer Canada Inc. Date of Revision: 17,300 Trans-Canada Highway 25 May 2009 Kirkland, Quebec, H9J 2M5 Control No. 120830 * TM Pharmacia Enterprises S.A. Pfizer Canada Inc., licensee © Pfizer Canada Inc., 2009 OGEN* (estropipate) Prescribing Information Page 1 of 27 Table of Contents PART I: HEALTH PROFESSIONAL INFORMATION.........................................................3 SUMMARY PRODUCT INFORMATION ........................................................................3 INDICATIONS AND CLINICAL USE..............................................................................3 CONTRAINDICATIONS ...................................................................................................3 WARNINGS AND PRECAUTIONS..................................................................................4 ADVERSE REACTIONS..................................................................................................11 CLINICAL TRIAL ADVERSE DRUG REACTIONS.....................................................13 DRUG INTERACTIONS ..................................................................................................13 DOSAGE AND ADMINISTRATION..............................................................................15 OVERDOSAGE ................................................................................................................16 ACTION AND CLINICAL PHARMACOLOGY ............................................................16 -

Postmenopausal Pharmacotherapy Newsletter
POSTMENOPAUSAL PHARMACOTHERAPY September, 1999 As Canada's baby boomers age, more and more women will face the option of Hormone Replacement Therapy (HRT). The HIGHLIGHTS decision can be a difficult one given the conflicting pros and cons. M This RxFiles examines the role and use of HRT, as well as newer Long term HRT carries several major benefits but also risks SERMS and bisphosphonates in post-menopausal (PM) patients. which should be evaluated on an individual and ongoing basis MContinuous ERT is appropriate for women without a uterus HRT MWomen with a uterus should receive progestagen (at least 12 HRT is indicated for the treatment of PM symptoms such as days per month or continuous low-dose) as part of their HRT vasomotor disturbances and urogenital atrophy, and is considered MLow-dose ERT (CEE 0.3mg) + Ca++ appears to prevent PMO primary therapy for prevention and treatment of postmenopausal MBisphosphinates (e.g. alendronate, etidronate) and raloxifene are osteoporosis (PMO).1 Contraindications are reviewed in Table 2. alternatives to HRT in treating and preventing PMO Although HRT is contraindicated in women with active breast or M"Natural" HRT regimens can be compounded but data is lacking uterine cancer, note that a prior or positive family history of these does not necessarily preclude women from receiving HRT.1 Comparative Safety: Because of differences between products, some side effects may be alleviated by switching from one product Estrogen Replacement Therapy (ERT) 2 to another, particularly from equine to plant sources or from oral to Naturally secreted estrogens include: topical (see Table 3 - Side Effects & Their Management). -

Optum Essential Health Benefits Enhanced Formulary PDL January
PENICILLINS ketorolac tromethamineQL GENERIC mefenamic acid amoxicillin/clavulanate potassium nabumetone amoxicillin/clavulanate potassium ER naproxen January 2016 ampicillin naproxen sodium ampicillin sodium naproxen sodium CR ESSENTIAL HEALTH BENEFITS ampicillin-sulbactam naproxen sodium ER ENHANCED PREFERRED DRUG LIST nafcillin sodium naproxen DR The Optum Preferred Drug List is a guide identifying oxacillin sodium oxaprozin preferred brand-name medicines within select penicillin G potassium piroxicam therapeutic categories. The Preferred Drug List may piperacillin sodium/ tazobactam sulindac not include all drugs covered by your prescription sodium tolmetin sodium drug benefit. Generic medicines are available within many of the therapeutic categories listed, in addition piperacillin sodium/tazobactam Fenoprofen Calcium sodium to categories not listed, and should be considered Meclofenamate Sodium piperacillin/tazobactam as the first line of prescribing. Tolmetin Sodium Amoxicillin/Clavulanate Potassium LOW COST GENERIC PREFERRED For benefit coverage or restrictions please check indomethacin your benefit plan document(s). This listing is revised Augmentin meloxicam periodically as new drugs and new prescribing LOW COST GENERIC naproxen kit information becomes available. It is recommended amoxicillin that you bring this list of medications when you or a dicloxacillin sodium CARDIOVASCULAR covered family member sees a physician or other penicillin v potassium ACE-INHIBITORS healthcare provider. GENERIC QUINOLONES captopril ANTI-INFECTIVES -

Pp375-430-Annex 1.Qxd
ANNEX 1 CHEMICAL AND PHYSICAL DATA ON COMPOUNDS USED IN COMBINED ESTROGEN–PROGESTOGEN CONTRACEPTIVES AND HORMONAL MENOPAUSAL THERAPY Annex 1 describes the chemical and physical data, technical products, trends in produc- tion by region and uses of estrogens and progestogens in combined estrogen–progestogen contraceptives and hormonal menopausal therapy. Estrogens and progestogens are listed separately in alphabetical order. Trade names for these compounds alone and in combination are given in Annexes 2–4. Sales are listed according to the regions designated by WHO. These are: Africa: Algeria, Angola, Benin, Botswana, Burkina Faso, Burundi, Cameroon, Cape Verde, Central African Republic, Chad, Comoros, Congo, Côte d'Ivoire, Democratic Republic of the Congo, Equatorial Guinea, Eritrea, Ethiopia, Gabon, Gambia, Ghana, Guinea, Guinea-Bissau, Kenya, Lesotho, Liberia, Madagascar, Malawi, Mali, Mauritania, Mauritius, Mozambique, Namibia, Niger, Nigeria, Rwanda, Sao Tome and Principe, Senegal, Seychelles, Sierra Leone, South Africa, Swaziland, Togo, Uganda, United Republic of Tanzania, Zambia and Zimbabwe America (North): Canada, Central America (Antigua and Barbuda, Bahamas, Barbados, Belize, Costa Rica, Cuba, Dominica, El Salvador, Grenada, Guatemala, Haiti, Honduras, Jamaica, Mexico, Nicaragua, Panama, Puerto Rico, Saint Kitts and Nevis, Saint Lucia, Saint Vincent and the Grenadines, Suriname, Trinidad and Tobago), United States of America America (South): Argentina, Bolivia, Brazil, Chile, Colombia, Dominican Republic, Ecuador, Guyana, Paraguay, -

Rheumatology: 15. Osteoporosis Review John P
Clinical basics Rheumatology: 15. Osteoporosis Review John P. Wade Synthèse The case Mrs. C is an otherwise healthy, 58-year-old woman who consults her primary Dr. Wade is with the Division care physician after experiencing the acute onset of severe low thoracic pain. of Rheumatology, University She reports having lifted her heavy grandson the previous day. X-ray films of of British Columbia, and the thoracic spine show a 50% compression fracture of the T11 vertebra. She Vancouver General Hospital, has always had a reasonable intake of calcium and maintains an active Vancouver, BC. lifestyle. She reached menopause at the age of 48 years, but she did not choose hormone replacement therapy because she was concerned that it This article has been peer reviewed. might be associated with breast cancer. She continues to smoke 1 pack of cig- arettes a day (despite counselling from her physician) and drinks alcohol so- CMAJ 2001;165(1):45-50 cially. She has no family history of osteoporosis or cardiac disease. Mrs. C This series has been reviewed had a hysterectomy 10 years ago for reasons that are unclear. She is accom- and endorsed by the Canadian panied by her only daughter, a nurse, who is anxious that the appropriate Rheumatology Association. tests be carried out and that treatment be started immediately. The Arthritis Society salutes CMAJ steoporosis is a common clinical problem that has been well reviewed.1–3 for its extensive series of articles The lifetime risk of an osteoporotic fracture is 1 in 4 among women and on arthritis. The Society believes 1 in 8 among men. -
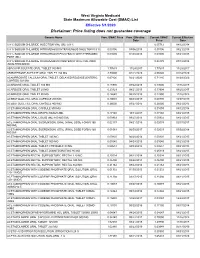
Disclaimer: Price Listing Does Not Guarantee Coverage
West Virginia Medicaid State Maximum Allowable Cost (SMAC) List Effective 5/1/2020 Disclaimer: Price listing does not guarantee coverage Generic Name Prev. SMAC Price Prev. Effective Current SMAC Current Effective Date Price Date 0.9 % SODIUM CHLORIDE INJECTION VIAL (ML) 0.9 % 0.05712 08/12/2014 0.9 % SODIUM CHLORIDE INTRAVENOUS INTRAVENOUS SOLUTION 0.9 % 0.00196 03/06/2015 0.00196 08/21/2015 0.9 % SODIUM CHLORIDE INTRAVENOUS PIGGYBACK WITH THREADED 0.03505 01/30/2015 0.03505 08/21/2015 PORT (ML) 0.9 % SODIUM CHLORIDE INTRAVENOUS PIGGYBACK WITH VIAL PORT 0.04175 07/18/2014 (NON-THREADED) ABACAVIR SULFATE ORAL TABLET 300 MG 1.77633 11/24/2017 1.77633 11/25/2017 ABIRATERONE ACETATE ORAL TABLET 250 MG 3.78000 02/21/2020 4.56040 02/24/2020 ACAMPROSATE CALCIUM ORAL TABLET, DELAYED RELEASE (ENTERIC 0.67426 02/21/2020 0.74145 04/09/2020 COATED) 333 MG ACARBOSE ORAL TABLET 100 MG 0.18990 03/02/2018 0.13000 11/16/2018 ACARBOSE ORAL TABLET 25 MG 0.23329 08/21/2015 0.13994 08/25/2017 ACARBOSE ORAL TABLET 50 MG 0.14640 05/25/2018 0.12500 11/16/2018 ACEBUTOLOL HCL ORAL CAPSULE 200 MG 0.40001 06/04/2019 0.60776 10/07/2019 ACEBUTOLOL HCL ORAL CAPSULE 400 MG 0.26000 03/27/2015 0.26000 08/21/2015 ACETAMINOPHEN ORAL CAPSULE 500 MG 0.01093 08/12/2014 ACETAMINOPHEN ORAL DROPS 80MG/0.8ML 0.17160 10/11/2013 0.17160 08/21/2015 ACETAMINOPHEN ORAL LIQUID (ML) 160 MG/5ML 0.00932 09/27/2013 0.00932 08/21/2015 ACETAMINOPHEN ORAL SUSPENSION, ORAL (FINAL DOSE FORM) 160 0.02137 08/21/2015 0.02033 02/17/2017 MG/5ML ACETAMINOPHEN ORAL SUSPENSION, ORAL (FINAL DOSE FORM) -

These Highlights Do Not Include All the Information Needed to Use PRASTERA® Safely and Effectively
PRASTERA- prasterone and ibuprofen Health Science Funding, LLC Disclaimer: This drug has not been found by FDA to be safe and effective, and this labeling has not been approved by FDA. For further information about unapproved drugs, click here. ---------- HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use PRASTERA® safely and effectively. See full prescribing information for PRASTERA® . PRASTERA® prasterone oral softgels 200mg are for oral use only. Initial U.S. Approval INDICATIONS AND USAGE PRASTERA® prasterone 200 mg oral softgels is indicated in female patients with mild to moderate, active (SLEDAI ≥2) systemic lupus erythematosus (SLE) to restore serum 5-dehydroandrosterone sulfate to levels typical of women without SLE. In Phase III clinical trials in female patients with mild to moderate active SLE, prasterone 200 mg was associated with reduced risk of auto-immune flare, reduced risk of breast cancer and reduced risk of death from any cause. (§§1, 6.2, 6.4, 6.5) DOSAGE AND ADMINISTRATION The recommended dose is one (1) softgel daily. (§2) DOSAGE FORMS AND STRENGTHS 200mg oral softgel capsules supplied in a convenience package with ibuprofen oral tablets 400mg. (§3) CONTRAINDICATIONS Known hypersensitivity to any of its ingredients. (§4) Undiagnosed abnormal genital bleeding. (§4) Known, suspected or history of breast cancer. (§4, §6.5) History of, or known, deep vein thrombosis, pulmonary embolism, arterial thromboembolic disease (e.g., stroke, myocardial infarction). (§4) Hypercholesterolemia or ischemic heart disease. (§4, §7.2.4) Hepatic or renal impairment (pharmacokinetic data lacking). (§4, §7.2) Breast-feeding or known or suspected pregnancy. -

OGEN® Estropipate Tablets, USP
PATIENT INFORMATION (Updated July 2006) OGEN® estropipate tablets, USP Read this PATIENT INFORMATION before you start taking OGEN and read what you get each time you refill OGEN. There may be new information. This information does not take the place of talking to your health care provider about your medical condition or your treatment. WHAT IS THE MOST IMPORTANT INFORMATION I SHOULD KNOW ABOUT OGEN (AN ESTROGEN HORMONE)? • Estrogens increase the chances of getting cancer of the uterus. Report any unusual vaginal bleeding right away while you are taking estrogens. Vaginal bleeding after menopause may be a warning sign of cancer of the uterine (womb). Your health care provider should check any unusual vaginal bleeding to find out the cause. • Do not use estrogens with or without progestins to prevent heart disease, heart attacks or strokes. Using estrogens with or without progestins may increase your chances of getting heart attacks, strokes, breast cancer and blood clots. You and your health care provider should talk regularly about whether you still need treatment with OGEN. What is OGEN? OGEN is a medicine that contains estrogen hormones. What is OGEN used for? OGEN is used during and after menopause to: • reduce moderate or severe hot flashes. Estrogens are hormones made by a woman’s ovaries. The ovaries normally stop making estrogens when a woman is between 45 to 55 years old. This drop in body estrogen levels causes the “change of life” or menopause (the end of monthly menstrual periods). Sometimes, both ovaries are removed during an operation before natural menopause takes place. -

DUAVEE® (Conjugated Estrogens-Bazedoxifene) Oral Tablet
PHARMACY COVERAGE GUIDELINES ORIGINAL EFFECTIVE DATE: 11/17/2016 SECTION: DRUGS LAST REVIEW DATE: 11/19/2020 LAST CRITERIA REVISION DATE: 11/19/2020 ARCHIVE DATE: DUAVEE® (conjugated estrogens-bazedoxifene) oral tablet Coverage for services, procedures, medical devices and drugs are dependent upon benefit eligibility as outlined in the member's specific benefit plan. This Pharmacy Coverage Guideline must be read in its entirety to determine coverage eligibility, if any. This Pharmacy Coverage Guideline provides information related to coverage determinations only and does not imply that a service or treatment is clinically appropriate or inappropriate. The provider and the member are responsible for all decisions regarding the appropriateness of care. Providers should provide BCBSAZ complete medical rationale when requesting any exceptions to these guidelines. The section identified as “Description” defines or describes a service, procedure, medical device or drug and is in no way intended as a statement of medical necessity and/or coverage. The section identified as “Criteria” defines criteria to determine whether a service, procedure, medical device or drug is considered medically necessary or experimental or investigational. State or federal mandates, e.g., FEP program, may dictate that any drug, device or biological product approved by the U.S. Food and Drug Administration (FDA) may not be considered experimental or investigational and thus the drug, device or biological product may be assessed only on the basis of medical necessity. Pharmacy Coverage Guidelines are subject to change as new information becomes available. For purposes of this Pharmacy Coverage Guideline, the terms "experimental" and "investigational" are considered to be interchangeable. -

Bioidentical Hormones: an Evidence-Based Review for Primary Care Providers
REVIEW Bioidentical Hormones: An Evidence-Based Review for Primary Care Providers Eileen Conaway, DO Context: Since 2002, when the US Food and Drug Adminis - combination with estrogen for the management of vasomotor tration (FDA) placed a black box warning on women’s hor - symptoms. Dehydroepiandrosterone is not FDA approved, mone replacement products, women and their providers but small-scale studies indicate it may improve bone min - have been struggling with whether to proceed with hormone eral density. Data are conflicting about efficacy in improving replacement therapy. Out of the controversy has grown a sexual dysfunction. There is an abundance of misleading popular movement promoting the use of bioidentical hor - information available in the media and on the Internet for mones. Many providers are still unsure if they want to rec - our patients. Compounded bioidenticals and salivary hor - ommend these products and, if so, how to use them appro - mone testing are unnecessary, are not standardized, and priately. should be avoided. Objective: To inform primary care providers (eg, physicians, Conclusion: Bioidentical hormones that are approved by the physician assistants, nurse practitioners) about current data on FDA may be preferred over standard hormone replacement the safety and efficacy of bioidentical hormone replacement because of their physiologic benefits and safety profile. therapy and to provide a context for patient perceptions. J Am Osteopath Assoc . 2011;111(3):153-164 Methods: Literature published between 1999 and 2009 was reviewed through MD Consult’s Medline and Ovid search n the aftermath of the unexpected adverse results of the engines. A Google search of popular media was also per - IWomen’s Health Initiative (WHI) trial 1 in 2002, women formed using the same terms. -

Recent Advances in the Analysis of Steroid Hormones and Related Drugs
ANALYTICAL SCIENCES MAY 2004, VOL. 20 767 2004 © The Japan Society for Analytical Chemistry Reviews Recent Advances in the Analysis of Steroid Hormones and Related Drugs Sándor GÖRÖG Gedeon Richter Ltd., P.O.B. 27, H-1475 Budapest, Hungary The development during the last 15 years and the state-of-the-art in the analysis of bulk steroid hormone drugs and hormone-like structures and pharmaceutical formulations made thereof are summarized. Other steroids (sterols, bile acids, cardiac glycosides, vitamins D) as well as biological-clinical aspects and pharmacokinetic and metabolic studies are excluded from this review. The state-of-the-art is summarized based on comparisons of monographs in the latest editions of the European Pharmacopoeia, United States Pharmacopoeia and the Japanese Pharmacopoeia. This is followed by sections dealing with new developments in the methodology for the fields of spectroscopic and spectrophotometric, chromatographic, electrophoretic and hyphenated techniques as well electroanalytical methods. The review is terminated by two problem-oriented sections: examples on impurity and degradation profiling as well as enantiomeric analysis. (Received January 14, 2004; Accepted February 2, 2004) 1 Introduction 767 4·3 Supercritical fluid chromatography (SFC) 2 Steroid Hormone Drugs in Pharmacopoeias 768 4·4 High-performance liquid chromatography 2·1 Assay of bulk drug materials (HPLC) and HPLC-MS 2·2 Related impurities test of bulk drug materials 5 Electrophoretic and Related Methods 776 2·3 Assay of steroid hormone formulations