Jaime Gómez Díaz
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
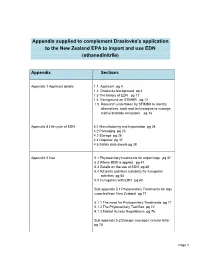
Appendix Supplied to Complement Draslovka's Application to the New
Appendix supplied to complement Draslovka’s application to the New Zealand EPA to import and use EDN (ethanedinitrile) Appendix Sections Appendix 1 Applicant details 1.1. Applicant pg 4 1.2 Draslovka background pg 4 1.3 The history of EDN pg 11 1.4 Background on STIMBR pg 12 1.5. Research undertaken by STIMBR to identify alternatives, tools and technologies to manage methyl bromide emissions pg 16 Appendix 4 Life cycle of EDN 4.1 Manufacturing and Importation pg 24 4.2 Packaging pg 25 4.3 Storage pg 28 4.4 Disposal pg 37 4.5 Safety data sheets pg 38 Appendix 5 Use 5.1 Phytosanitary treatments for export logs pg 47 5.2 Where EDN is applied pg 47 5.3 Details on the use of EDN pg 48 5.4 NZ ports and their suitability for fumigation activities pg 50 5.5 Fumigation with EDN pg 60 Sub appendix 5.1 Phytosanitary Treatments for logs exported from New Zealand pg 71 5.1.1 The need for Phytosanitary Treatments pg 71 5.1.2 The Phytosanitary Tool Box pg 72 5.1.3 Market Access Negotiations pg 76 Sub appendix 5.2 Draeger cyanogen circular letter pg 78 Page 1 Appendix 6 Toxicology and HSNO 6.1 Classification pg 79 hazard classifications of the formulated 6.2 Toxicology pg 87 substance 6.2.1 Mode of action and toxic kinetic pg 87 6.2.2 Acute toxicity pg 88 Acute inhalation Oral toxicity Dermal toxicity Eye irritancy 6.2.3 Systemic toxicity pg 90 6.2.4 Reproductive and developmental toxicity pg 90 6.2.5 Neurotoxicity pg 91 6.2.6 Chronic toxicity pg 92 6.2.7 Genotoxicity pg 93 6.2.8 Carcinogenicity pg 95 6.2.9 WES and TEL values pg 96 6.3 Eco-toxicology 6.3.1 Fate in the environment pg 111 6.3.2 Ecotoxicological Classifications pg 113 Appendix 7 7.2 Risks costs and benefits analysis of 7.2.1 Risks pg 114 Introduction Proposed use Product description: and approach to risk analysis Physical characteristic assessment Human toxicological assessment Ecotoxicology risk assessment 7.2.2 Costs pg 137 Associated with a new chemical. -
Cyanide Process
Cyanide Process The cyanide antidote kit consists of three medications given together: amyl nitrite, sodium nitrite, and sodium thiosulfate. Sodium cyanide solution in water is a strong base; it reacts violently with acid and is corrosive. How is cyanide waste treated at your facility? Chemically speaking it looks like this CN + 2H20 -> NH3 + HCOO in laymen's terms we destroy cyanide using Thermal Hydrolysis. Liquid – vapor diagram of the system H 2O– HCN at atmospheric. The process was invented in 1887 by the Scottish chemists John S. 1 word related to cyanide process: industrial process. net dictionary. txt) or read online for free. Separate the process into half reactions.. 7k points). Australia is a major global manufacturer and exporter of sodium cyanide, with manufacturing facilities in western Australia. Orica's world leading cyanide analysers have been designed to optimise cyanide usage and lower costs, whilst maintaining precious metal recovery rates. Cyanide in fruits and vegetables is in the form of cyanogenic glycosides (cyanoglycosides). A cyanide is a compound, known simply as a 'cyanide', that contains a cyanide group (CN-) 1,3-6. After the door is sealed, and when the warden gives the signal, the executioner in a separate room operates a lever that releases the cyanide into the liquid. Whilst hanging is one of the most reliable methods of suicide, as for firearms, it is not 100% effective - studies would suggest 77% - 88% effective 1. 1 word related to cyanide process: industrial process. For the gold concentrate cyanide plant, the CaO concentration of the general control leaching process is between 2 and 5/10,000. -

The Structure of Catalyst Gauzes After Hydrogen Cyanide Production
The Structure of Catalyst Gauzes J after Hydrogen Cyanide Production A POSSIBLE EXPLANATION FOR THE FACETED FEATURES By A. G. Knapton Group Research Centre, Johnson Matthey and Co Limited During hydrogen cyanide production by the Andrussow process con- siderable redistribution of the surface of the rhodium-platinum gauze catalyst occurs, resulting in the formation of a porous mass of well-faceted metal crystallites. The phenomenon is illustrated in this paper, and some possible mechanisms to account for its formation are discussed. Rhodium-platinum alloy gauzes feature surface that occurs during operation. Much prominently in two important manufacturing has already been written in this journal (3, processes, the catalytic oxidation of ammonia 4, 5) on the surface effects on gauzes used for to nitric oxide in the production of nitric ammonia oxidation, but little attention has acid, and the Andrussow process for making been paid to gauze packs used in hydrogen hydrogen cyanide from methane and ammonia. cyanide production. At the Johnson Matthey Although direct reaction of the latter con- Research Centre we have recently had occa- stituents is possible via: sion to examine in detail gauzes that had been used in long campaigns in the Andrussow CH4 + NH, + HCN -1 3H, 4 100 kcal process. These gauzes showed some unusual and has been employed on a large scale (I), features, not found in those from ammonia the reaction is strongly endothermic, and oxidation and which have not been satisfac- supplying the heat to maintain the reaction torily explained by earlier workers (6, 7, 8, 9) temperature of 1000 to 1200T makes it on rhodium-platinum catalysts.