NIH Public Access Author Manuscript Curr Opin Virol
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cytokine Nomenclature
RayBiotech, Inc. The protein array pioneer company Cytokine Nomenclature Cytokine Name Official Full Name Genbank Related Names Symbol 4-1BB TNFRSF Tumor necrosis factor NP_001552 CD137, ILA, 4-1BB ligand receptor 9 receptor superfamily .2. member 9 6Ckine CCL21 6-Cysteine Chemokine NM_002989 Small-inducible cytokine A21, Beta chemokine exodus-2, Secondary lymphoid-tissue chemokine, SLC, SCYA21 ACE ACE Angiotensin-converting NP_000780 CD143, DCP, DCP1 enzyme .1. NP_690043 .1. ACE-2 ACE2 Angiotensin-converting NP_068576 ACE-related carboxypeptidase, enzyme 2 .1 Angiotensin-converting enzyme homolog ACTH ACTH Adrenocorticotropic NP_000930 POMC, Pro-opiomelanocortin, hormone .1. Corticotropin-lipotropin, NPP, NP_001030 Melanotropin gamma, Gamma- 333.1 MSH, Potential peptide, Corticotropin, Melanotropin alpha, Alpha-MSH, Corticotropin-like intermediary peptide, CLIP, Lipotropin beta, Beta-LPH, Lipotropin gamma, Gamma-LPH, Melanotropin beta, Beta-MSH, Beta-endorphin, Met-enkephalin ACTHR ACTHR Adrenocorticotropic NP_000520 Melanocortin receptor 2, MC2-R hormone receptor .1 Activin A INHBA Activin A NM_002192 Activin beta-A chain, Erythroid differentiation protein, EDF, INHBA Activin B INHBB Activin B NM_002193 Inhibin beta B chain, Activin beta-B chain Activin C INHBC Activin C NM005538 Inhibin, beta C Activin RIA ACVR1 Activin receptor type-1 NM_001105 Activin receptor type I, ACTR-I, Serine/threonine-protein kinase receptor R1, SKR1, Activin receptor-like kinase 2, ALK-2, TGF-B superfamily receptor type I, TSR-I, ACVRLK2 Activin RIB ACVR1B -

Overlapping, Additive and Counterregulatory Effects of Type II and I Interferons on Myeloid Dendritic Cell Functions
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Serveur académique lausannois Review Article TheScientificWorldJOURNAL (2011) 11, 2071–2090 ISSN 1537-744X; doi:10.1100/2011/873895 Overlapping, Additive and Counterregulatory Effects of Type II and I Interferons on Myeloid Dendritic Cell Functions Loredana Frasca and Roberto Lande Department of Infectious, Parasitic and Immune-mediated Diseases, Istituto Superiore di Sanita,` 00161 Rome, Italy Received 8 April 2011; Accepted 27 September 2011 Academic Editor: Fulvio D’Acquisto Dendritic cells (DCs) are central player in immunity by bridging the innate and adaptive arms of the immune system (IS). Interferons (IFNs) are one of the most important factors that regulate both innate and adaptive immunity too. Thus, the understanding of how type II and I IFNs modulate the immune-regulatory properties of DCs is a central issue in immunology. In this paper, we will address this point in the light of the most recent literature, also highlighting the controversial data reported in the field. According to the wide literature available, type II as well as type I IFNs appear, at the same time, to collaborate, to induce additive effects or overlapping functions, as well as to counterregulate each one’s effects on DC biology and, in general, the immune response. The knowledge of these effects has important therapeutic implications in the treatment of infec- tious/autoimmune diseases and cancer and indicates strategies for using IFNs as vaccine adju- vants and in DC-based immune therapeutic approaches. KEYWORDS: IFNs, dendritic cell effector functions, immune activation, immune regulation, cross- regulation Correspondence should be addressed to Loredana Frasca, [email protected] Copyright © 2011 L. -

Cyclooxygenase-2 Inflammatory Factors, IL-29, IL-8, And
Hepatitis B Virus Induces a Novel Inflammation Network Involving Three Inflammatory Factors, IL-29, IL-8, and Cyclooxygenase-2 This information is current as of September 27, 2021. Yi Yu, Rui Gong, Yongxin Mu, Yanni Chen, Chengliang Zhu, Zhichen Sun, Mingzhou Chen, Yingle Liu, Ying Zhu and Jianguo Wu J Immunol 2011; 187:4844-4860; Prepublished online 28 September 2011; Downloaded from doi: 10.4049/jimmunol.1100998 http://www.jimmunol.org/content/187/9/4844 http://www.jimmunol.org/ References This article cites 55 articles, 29 of which you can access for free at: http://www.jimmunol.org/content/187/9/4844.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 27, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2011 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Hepatitis B Virus Induces a Novel Inflammation Network Involving Three Inflammatory Factors, IL-29, IL-8, and Cyclooxygenase-2 Yi Yu,*,†,‡ Rui Gong,*,† Yongxin Mu,*,† Yanni Chen,*,†,‡ Chengliang Zhu,*,† Zhichen Sun,*,† Mingzhou Chen,*,†,‡ Yingle Liu,*,†,‡ Ying Zhu,*,†,‡ and Jianguo Wu*,†,‡ Chronic inflammation induced by hepatitis B virus (HBV) is a major causative factor associated with the development of cirrhosis and hepatocellular carcinoma. -

HUMAN INTERLEUKIN-29/INTERFERON-LAMBDA 1 Product Number: 11825-1 Lot Number: 5620 Expiration Date: October 2, 2013 Size: 25 Μg
HUMAN INTERLEUKIN-29/INTERFERON-LAMBDA 1 Product Number: 11825-1 Lot Number: 5620 Expiration Date: October 2, 2013 Size: 25 μg Description: Human Interleukin-29/Interferon-Lambda 1 Source: DNA sequence encoding the signal peptide from human CD33 was fused to the carboxyl terminally polyhistidine-tagged mature human IL-29 (Gly 20 - Thr 200) (Sheppard, P. et al., 2003, Nat. Immunol. 4(1):63 - 68). The chimeric protein was expressed in a mouse myeloma cell line, NS0. Form: Lyophilized Buffer: Phosphate-buffered saline (PBS) containing 50μg of bovine serum albumin (BSA) per 1 μg of cytokine Reconstitution: It is recommended that sterile PBS containing at least 0.1% human serum albumin or bovine serum albumin be added to the vial to prepare a stock solution of no less than 10μg/ml. Endotoxin: < 1 EU/μg Molecular Weight: Based on N-terminal sequencing, the mature recombinant IL-29 starts at Gly 20 and has a calculated molecular mass of 21.4 kDa. As a result of glycosylation, the recombinant monomer migrates as an approximately 26-35kDa protein in SDS-PAGE under reducing conditions. Purity: > 95% Synonyms: Hu IL-29; Hu IFN-λ1 Assays Used to Measure Bioactivity: Human HepG2 cells infected with encephalomyocarditis virus (Sheppard, P. et al., 2003, Nature Immunol. 4:63). The ED50 for this effect is typically 1- 5 ng/mL. Shipping Conditions: Wet Ice Physical State of Product During Shipping: Lyophilized Special Conditions/Comments: After receipt, this product should be kept at -70˚C or below for retention of full activity. Upon reconstitution, this cytokine can be stored under sterile conditions at -20˚C to -70˚C in a manual defrost freezer for three months without detection loss of activity. -

Cells 1/IL-29) in Human Airway Epithelial Λ (IFN- 1 Promoter Activity
Regulation of IFN-λ1 Promoter Activity (IFN- λ1/IL-29) in Human Airway Epithelial Cells This information is current as Rachael Siegel, Joyce Eskdale and Grant Gallagher of September 27, 2021. J Immunol 2011; 187:5636-5644; Prepublished online 4 November 2011; doi: 10.4049/jimmunol.1003988 http://www.jimmunol.org/content/187/11/5636 Downloaded from References This article cites 48 articles, 19 of which you can access for free at: http://www.jimmunol.org/content/187/11/5636.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 27, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2011 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Regulation of IFN-l1 Promoter Activity (IFN-l1/IL-29) in Human Airway Epithelial Cells Rachael Siegel, Joyce Eskdale, and Grant Gallagher The type III (l) IFNs (IFN-l1, IFN-l2, and IFN-l3) and their receptor are the most recently discovered IFN family. -

Secreted Factors from Dental Pulp Stem Cells Improve Sjögren's Syndrome Via Regulatory T Cell-Mediated Immunosuppression
Matsumura-Kawashima et al. Stem Cell Research & Therapy (2021) 12:182 https://doi.org/10.1186/s13287-021-02236-6 RESEARCH Open Access Secreted factors from dental pulp stem cells improve Sjögren’s syndrome via regulatory T cell-mediated immunosuppression Mayu Matsumura-Kawashima†, Kenichi Ogata*† , Masafumi Moriyama, Yuka Murakami, Tatsuya Kawado and Seiji Nakamura Abstract Background: Sjögren’s syndrome (SS) is a chronic autoimmune disease primarily characterized by inflammation in the salivary and lacrimal glands. Activated T cells contribute to disease pathogenesis by producing proinflammatory cytokines, which leads to a positive feedback loop establishment. The study aimed to evaluate the effects of secreted factors derived from dental pulp stem cells (DPSCs) or bone marrow mesenchymal stem cells (BMMSCs) on hyposalivation in SS and to investigate the mechanism involved. Methods: Eighty percent confluent stem cells were replenished with serum-free Dulbecco’s modified Eagle’s medium and incubated for 48 h; following which, conditioned media from DPSCs (DPSC-CM) and BMMSCs (BMMSC-CM) were collected. Cytokine array analysis was performed to assess the types of cytokines present in the media. Flow cytometric analysis was performed to evaluate the number of activated T cells cultured in DPSC-CM or BMMSC-CM. Subsequently, DPSC-CM or BMMSC-CM was administered to an SS mouse model. The mice were categorized into the following groups (n = 6 each): non-treatment, Dulbecco’s modified Eagle’s medium (−), BMMSC-CM, and DPSC-CM. Histological analysis of the salivary glands was performed. The gene and protein expression levels of cytokines associated with T helper subsets in the submandibular glands (SMGs) were evaluated. -

The Role of Il-29 and Il-28B in the Innate Immune Response
THE ROLE OF IL-29 AND IL-28B IN THE INNATE IMMUNE RESPONSE Megumi A. Williamson A thesis submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Master of Science in the Department of Periodontology, School of Dentistry Chapel Hill 2018 Approved by: Thiago Morelli Thiago Morelli Julie Marchesan Steven Offenbacher Antonio Amelio ©2018 Megumi A. Williamson ALL RIGHTS RESERVED ii ABSTRACT Megumi A. Williamson: The Role of IL-29 and IL-28B in the Innate Immune Response (Under the direction of Thiago Morelli, Julie Marchesan, Steven Offenbacher, and Antonio Amelio) Aims: Chronic periodontitis (CP) is an inflammatory disease induced by dysbiotic biofilm in a susceptible host, resulting in progressive attachment loss, and subsequent alveolar bone loss. Recent genome-wide association studies (GWAS) and genome-wide gene centric analysis on periodontal complex traits (PCTs) identified possible associations of IL-29 and IL- 28B with periodontal diseases. However, the underlying mechanisms for how these genes contribute to the pathogenesis of periodontitis are largely unknown. The aims of the present study were to explore the role of IL-29 and IL-28B and their gene polymorphisms in the innate immune response by dendritic cells. Materials and methods: To explore the effect of IL-29 on the cytokine production in response to TLR4 stimulation, the IL-29 gene was knocked-down in THP-1 cells using IL-29 shRNA lentiviral particles. Pro- and anti-inflammatory cytokine levels were measured with Luminex® multiplex assay. To assess the effect of genetic variations in IL- 29 and IL-28B, whole blood samples from fifteen subjects (6 subjects with major allele for both IL-29 and IL-28B, 5 subjects for major allele in IL-29 and minor allele in IL-28B, and 4 subjects with minor alleles for both genes) were collected and CD14+/CD16lo PBMCs were isolated to generate DCs. -

Evolutionary Divergence and Functions of the Human Interleukin (IL) Gene Family Chad Brocker,1 David Thompson,2 Akiko Matsumoto,1 Daniel W
UPDATE ON GENE COMPLETIONS AND ANNOTATIONS Evolutionary divergence and functions of the human interleukin (IL) gene family Chad Brocker,1 David Thompson,2 Akiko Matsumoto,1 Daniel W. Nebert3* and Vasilis Vasiliou1 1Molecular Toxicology and Environmental Health Sciences Program, Department of Pharmaceutical Sciences, University of Colorado Denver, Aurora, CO 80045, USA 2Department of Clinical Pharmacy, University of Colorado Denver, Aurora, CO 80045, USA 3Department of Environmental Health and Center for Environmental Genetics (CEG), University of Cincinnati Medical Center, Cincinnati, OH 45267–0056, USA *Correspondence to: Tel: þ1 513 821 4664; Fax: þ1 513 558 0925; E-mail: [email protected]; [email protected] Date received (in revised form): 22nd September 2010 Abstract Cytokines play a very important role in nearly all aspects of inflammation and immunity. The term ‘interleukin’ (IL) has been used to describe a group of cytokines with complex immunomodulatory functions — including cell proliferation, maturation, migration and adhesion. These cytokines also play an important role in immune cell differentiation and activation. Determining the exact function of a particular cytokine is complicated by the influence of the producing cell type, the responding cell type and the phase of the immune response. ILs can also have pro- and anti-inflammatory effects, further complicating their characterisation. These molecules are under constant pressure to evolve due to continual competition between the host’s immune system and infecting organisms; as such, ILs have undergone significant evolution. This has resulted in little amino acid conservation between orthologous proteins, which further complicates the gene family organisation. Within the literature there are a number of overlapping nomenclature and classification systems derived from biological function, receptor-binding properties and originating cell type. -
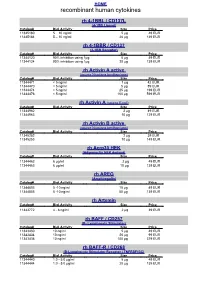
Recombinant Human Cytokines
HOME recombinant human cytokines rh 4-1BBL / CD137L (4-1BB Ligand) Catalog# Biol.Activity Size Price 11345180 5 – 10 ng/ml 5 µg 49 EUR 11345184 5 – 10 ng/ml 20 µg 139 EUR rh 4-1BBR / CD137 (4-1BB Receptor) Catalog# Biol.Activity Size Price 11344120 90% inhibition using 1µg 5 µg 49 EUR 11344124 90% inhibition using 1µg 20 µg 139 EUR rh Activin A active (source Nicotiana benthamiana) Catalog# Biol.Activity Size Price 11344471 < 5 ng/ml 1 µg 42 EUR 11344470 < 5 ng/ml 5 µg 89 EUR 11344474 < 5 ng/ml 25 µg 199 EUR 11344476 < 5 ng/ml 100 µg 599 EUR rh Activin A (source E.coli) Catalog# Biol.Activity Size Price 11344962 2 µg 49 EUR 11344963 10 µg 129 EUR rh Activin B active (source Nicotiana benthamiana) Catalog# Biol.Activity Size Price 11345252 2 µg 39 EUR 11345253 10 µg 149 EUR rh Acrp30 HEK (Adiponectin HEK derived) Catalog# Biol.Activity Size Price 11344462 6 µg/ml 2 µg 49 EUR 11344463 6 µg/ml 10 µg 139 EUR rh AREG (Amphiregulin) Catalog# Biol.Activity Size Price 11344803 5 -10 ng/ml 10 µg 49 EUR 11344805 5 -10 ng/ml 50 µg 139 EUR rh Artemin Catalog# Biol.Activity Size Price 11343772 4 - 8 ng/ml 2 µg 39 EUR rh BAFF / CD257 (B- Lymphocyte Stimulator) Catalog# Biol.Activity Size Price 11343430 10 ng/ml 5 µg 49 EUR 11343434 10 ng/ml 20 µg 99 EUR 11343436 10 ng/ml 100 µg 379 EUR rh BAFF-R / CD268 (B-Lymphocyte Stimulator Receptor / TNFRSF13C) Catalog# Biol.Activity Size Price 11344440 1.0 - 5.0 µg/ml 5 µg 49 EUR 11344444 1.0 - 5.0 µg/ml 20 µg 139 EUR HOME recombinant human cytokines rh BCA-1 (CXCL13) Catalog# Biol.Activity Size Price 11344180 -

Full-Text.Pdf
IL-17A Attenuates IFN-λ Expression by Inducing Suppressor of Cytokine Signaling Expression in Airway Epithelium This information is current as Mitsuru Niwa, Tomoyuki Fujisawa, Kazutaka Mori, of September 28, 2021. Katsumasa Yamanaka, Hideki Yasui, Yuzo Suzuki, Masato Karayama, Hironao Hozumi, Kazuki Furuhashi, Noriyuki Enomoto, Yutaro Nakamura, Naoki Inui, Tetsuro Suzuki, Masato Maekawa and Takafumi Suda J Immunol published online 17 September 2018 Downloaded from http://www.jimmunol.org/content/early/2018/09/14/jimmun ol.1800147 Supplementary http://www.jimmunol.org/content/suppl/2018/09/14/jimmunol.180014 http://www.jimmunol.org/ Material 7.DCSupplemental Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 28, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2018 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published -

(12) United States Patent (10) Patent No.: US 8.470,295 B2 Warren Et Al
USOO8470295B2 (12) United States Patent (10) Patent No.: US 8.470,295 B2 Warren et al. (45) Date of Patent: Jun. 25, 2013 (54) METHODS OF TREATMENT OF 6,486,146 B1 * 1 1/2002 Zamoyski ..................... 514,177 ANDROGENIC STEROIDAL, HORMONE 93. R 838. S. DEPENDENT CANCER WITH AUGER 6.658, 3.568. Seal ELECTRON-EMITTING NUCLEOSDE 2001/0007933 A1 7, 2001 Lesh et al. ANALOGS 2001/OOO997O A1 7/2001 Chornenky et al. 2001/0031941 A1 10, 2001 Edwards et al. (75) Inventors: Stephen L. Warren, Fort Collins, CO 38:9: A. &39: they al. (US), lames E. Matsura. Fort Collins, 2002/O123719 A1 9, 2002 Laviang et etal. al. CO (US); Michael J. Gerber, Denver, 2002/0133057 A1 9, 2002 Kukuk CO (US) 2002/0133173 A1 9, 2002 Brocket al. 2003/0028147 A1 2/2003 Aves et al. (73) Assignee: Peak Biosciences, Inc., Fort Collins, CO 2003/00931.17 A1 5.2003 Saadat (US) 2003.0167031 A1 9, 2003 Odland 2003/0171738 A1 9/2003 Konieczynski et al. 2004/022060.6 A1 11/2004 Goshgari (*) Notice: Subject to any disclaimer, the term of this 2004/02431.45 A1 12, 2004 SSan patent is extended or adjusted under 35 2005/0069495 A1* 3/2005 Baranowska-Kortylewicz U.S.C. 154(b) by 238 days. et al. ............................ 424,173 2005/0O85715 A1 4/2005 Dukesherer et al. (21) Appl. No.: 12/S99594 2005/0101823 A1 5/2005 Linares et al. 9 2005/O107738 A1 5/2005 Slater et al. 2005.0245858 A1 11/2005 Miesel et al. (22) PCT Filed: May 9, 2008 2006/O121085 A1 6/2006 Warren et al. -

Human Cytokine Response Profiles
Comprehensive Understanding of the Human Cytokine Response Profiles A. Background The current project aims to collect datasets profiling gene expression patterns of human cytokine treatment response from the NCBI GEO and EBI ArrayExpress databases. The Framework for Data Curation already hosted a list of candidate datasets. You will read the study design and sample annotations to select the relevant datasets and label the sample conditions to enable automatic analysis. If you want to build a new data collection project for your topic of interest instead of working on our existing cytokine project, please read section D. We will explain the cytokine project’s configurations to give you an example on creating your curation task. A.1. Cytokine Cytokines are a broad category of small proteins mediating cell signaling. Many cell types can release cytokines and receive cytokines from other producers through receptors on the cell surface. Despite some overlap in the literature terminology, we exclude chemokines, hormones, or growth factors, which are also essential cell signaling molecules. Meanwhile, we count two cytokines in the same family as the same if they share the same receptors. In this project, we will focus on the following families and use the member symbols as standard names (Table 1). Family Members (use these symbols as standard cytokine names) Colony-stimulating factor GCSF, GMCSF, MCSF Interferon IFNA, IFNB, IFNG Interleukin IL1, IL1RA, IL2, IL3, IL4, IL5, IL6, IL7, IL9, IL10, IL11, IL12, IL13, IL15, IL16, IL17, IL18, IL19, IL20, IL21, IL22, IL23, IL24, IL25, IL26, IL27, IL28, IL29, IL30, IL31, IL32, IL33, IL34, IL35, IL36, IL36RA, IL37, TSLP, LIF, OSM Tumor necrosis factor TNFA, LTA, LTB, CD40L, FASL, CD27L, CD30L, 41BBL, TRAIL, OPGL, APRIL, LIGHT, TWEAK, BAFF Unassigned TGFB, MIF Table 1.