Combined HIV-1 Sequence and Integration Site Analysis Informs Viral Dynamics and Allows Reconstruction of Replicating Viral Ancestors
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Deregulated Gene Expression Pathways in Myelodysplastic Syndrome Hematopoietic Stem Cells
Leukemia (2010) 24, 756–764 & 2010 Macmillan Publishers Limited All rights reserved 0887-6924/10 $32.00 www.nature.com/leu ORIGINAL ARTICLE Deregulated gene expression pathways in myelodysplastic syndrome hematopoietic stem cells A Pellagatti1, M Cazzola2, A Giagounidis3, J Perry1, L Malcovati2, MG Della Porta2,MJa¨dersten4, S Killick5, A Verma6, CJ Norbury7, E Hellstro¨m-Lindberg4, JS Wainscoat1 and J Boultwood1 1LRF Molecular Haematology Unit, NDCLS, John Radcliffe Hospital, Oxford, UK; 2Department of Hematology Oncology, University of Pavia Medical School, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy; 3Medizinische Klinik II, St Johannes Hospital, Duisburg, Germany; 4Division of Hematology, Department of Medicine, Karolinska Institutet, Stockholm, Sweden; 5Department of Haematology, Royal Bournemouth Hospital, Bournemouth, UK; 6Albert Einstein College of Medicine, Bronx, NY, USA and 7Sir William Dunn School of Pathology, University of Oxford, Oxford, UK To gain insight into the molecular pathogenesis of the the World Health Organization.6,7 Patients with refractory myelodysplastic syndromes (MDS), we performed global gene anemia (RA) with or without ringed sideroblasts, according to expression profiling and pathway analysis on the hemato- poietic stem cells (HSC) of 183 MDS patients as compared with the the French–American–British classification, were subdivided HSC of 17 healthy controls. The most significantly deregulated based on the presence or absence of multilineage dysplasia. In pathways in MDS include interferon signaling, thrombopoietin addition, patients with RA with excess blasts (RAEB) were signaling and the Wnt pathways. Among the most signifi- subdivided into two categories, RAEB1 and RAEB2, based on the cantly deregulated gene pathways in early MDS are immuno- percentage of bone marrow blasts. -

Mechanical Forces Induce an Asthma Gene Signature in Healthy Airway Epithelial Cells Ayşe Kılıç1,10, Asher Ameli1,2,10, Jin-Ah Park3,10, Alvin T
www.nature.com/scientificreports OPEN Mechanical forces induce an asthma gene signature in healthy airway epithelial cells Ayşe Kılıç1,10, Asher Ameli1,2,10, Jin-Ah Park3,10, Alvin T. Kho4, Kelan Tantisira1, Marc Santolini 1,5, Feixiong Cheng6,7,8, Jennifer A. Mitchel3, Maureen McGill3, Michael J. O’Sullivan3, Margherita De Marzio1,3, Amitabh Sharma1, Scott H. Randell9, Jefrey M. Drazen3, Jefrey J. Fredberg3 & Scott T. Weiss1,3* Bronchospasm compresses the bronchial epithelium, and this compressive stress has been implicated in asthma pathogenesis. However, the molecular mechanisms by which this compressive stress alters pathways relevant to disease are not well understood. Using air-liquid interface cultures of primary human bronchial epithelial cells derived from non-asthmatic donors and asthmatic donors, we applied a compressive stress and then used a network approach to map resulting changes in the molecular interactome. In cells from non-asthmatic donors, compression by itself was sufcient to induce infammatory, late repair, and fbrotic pathways. Remarkably, this molecular profle of non-asthmatic cells after compression recapitulated the profle of asthmatic cells before compression. Together, these results show that even in the absence of any infammatory stimulus, mechanical compression alone is sufcient to induce an asthma-like molecular signature. Bronchial epithelial cells (BECs) form a physical barrier that protects pulmonary airways from inhaled irritants and invading pathogens1,2. Moreover, environmental stimuli such as allergens, pollutants and viruses can induce constriction of the airways3 and thereby expose the bronchial epithelium to compressive mechanical stress. In BECs, this compressive stress induces structural, biophysical, as well as molecular changes4,5, that interact with nearby mesenchyme6 to cause epithelial layer unjamming1, shedding of soluble factors, production of matrix proteins, and activation matrix modifying enzymes, which then act to coordinate infammatory and remodeling processes4,7–10. -

Anti-ARL4A Antibody (ARG41291)
Product datasheet [email protected] ARG41291 Package: 100 μl anti-ARL4A antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes ARL4A Tested Reactivity Hu, Ms, Rat Tested Application ICC/IF, IHC-P Host Rabbit Clonality Polyclonal Isotype IgG Target Name ARL4A Antigen Species Human Immunogen Recombinant fusion protein corresponding to aa. 121-200 of Human ARL4A (NP_001032241.1). Conjugation Un-conjugated Alternate Names ARL4; ADP-ribosylation factor-like protein 4A Application Instructions Application table Application Dilution ICC/IF 1:50 - 1:200 IHC-P 1:50 - 1:200 Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Calculated Mw 23 kDa Properties Form Liquid Purification Affinity purified. Buffer PBS (pH 7.3), 0.02% Sodium azide and 50% Glycerol. Preservative 0.02% Sodium azide Stabilizer 50% Glycerol Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. Note For laboratory research only, not for drug, diagnostic or other use. www.arigobio.com 1/2 Bioinformation Gene Symbol ARL4A Gene Full Name ADP-ribosylation factor-like 4A Background ADP-ribosylation factor-like 4A is a member of the ADP-ribosylation factor family of GTP-binding proteins. ARL4A is similar to ARL4C and ARL4D and each has a nuclear localization signal and an unusually high guaninine nucleotide exchange rate. -

Defining Functional Interactions During Biogenesis of Epithelial Junctions
ARTICLE Received 11 Dec 2015 | Accepted 13 Oct 2016 | Published 6 Dec 2016 | Updated 5 Jan 2017 DOI: 10.1038/ncomms13542 OPEN Defining functional interactions during biogenesis of epithelial junctions J.C. Erasmus1,*, S. Bruche1,*,w, L. Pizarro1,2,*, N. Maimari1,3,*, T. Poggioli1,w, C. Tomlinson4,J.Lees5, I. Zalivina1,w, A. Wheeler1,w, A. Alberts6, A. Russo2 & V.M.M. Braga1 In spite of extensive recent progress, a comprehensive understanding of how actin cytoskeleton remodelling supports stable junctions remains to be established. Here we design a platform that integrates actin functions with optimized phenotypic clustering and identify new cytoskeletal proteins, their functional hierarchy and pathways that modulate E-cadherin adhesion. Depletion of EEF1A, an actin bundling protein, increases E-cadherin levels at junctions without a corresponding reinforcement of cell–cell contacts. This unexpected result reflects a more dynamic and mobile junctional actin in EEF1A-depleted cells. A partner for EEF1A in cadherin contact maintenance is the formin DIAPH2, which interacts with EEF1A. In contrast, depletion of either the endocytic regulator TRIP10 or the Rho GTPase activator VAV2 reduces E-cadherin levels at junctions. TRIP10 binds to and requires VAV2 function for its junctional localization. Overall, we present new conceptual insights on junction stabilization, which integrate known and novel pathways with impact for epithelial morphogenesis, homeostasis and diseases. 1 National Heart and Lung Institute, Faculty of Medicine, Imperial College London, London SW7 2AZ, UK. 2 Computing Department, Imperial College London, London SW7 2AZ, UK. 3 Bioengineering Department, Faculty of Engineering, Imperial College London, London SW7 2AZ, UK. 4 Department of Surgery & Cancer, Faculty of Medicine, Imperial College London, London SW7 2AZ, UK. -

Autism Multiplex Family with 16P11.2P12.2 Microduplication Syndrome in Monozygotic Twins and Distal 16P11.2 Deletion in Their Brother
European Journal of Human Genetics (2012) 20, 540–546 & 2012 Macmillan Publishers Limited All rights reserved 1018-4813/12 www.nature.com/ejhg ARTICLE Autism multiplex family with 16p11.2p12.2 microduplication syndrome in monozygotic twins and distal 16p11.2 deletion in their brother Anne-Claude Tabet1,2,3,4, Marion Pilorge2,3,4, Richard Delorme5,6,Fre´de´rique Amsellem5,6, Jean-Marc Pinard7, Marion Leboyer6,8,9, Alain Verloes10, Brigitte Benzacken1,11,12 and Catalina Betancur*,2,3,4 The pericentromeric region of chromosome 16p is rich in segmental duplications that predispose to rearrangements through non-allelic homologous recombination. Several recurrent copy number variations have been described recently in chromosome 16p. 16p11.2 rearrangements (29.5–30.1 Mb) are associated with autism, intellectual disability (ID) and other neurodevelopmental disorders. Another recognizable but less common microdeletion syndrome in 16p11.2p12.2 (21.4 to 28.5–30.1 Mb) has been described in six individuals with ID, whereas apparently reciprocal duplications, studied by standard cytogenetic and fluorescence in situ hybridization techniques, have been reported in three patients with autism spectrum disorders. Here, we report a multiplex family with three boys affected with autism, including two monozygotic twins carrying a de novo 16p11.2p12.2 duplication of 8.95 Mb (21.28–30.23 Mb) characterized by single-nucleotide polymorphism array, encompassing both the 16p11.2 and 16p11.2p12.2 regions. The twins exhibited autism, severe ID, and dysmorphic features, including a triangular face, deep-set eyes, large and prominent nasal bridge, and tall, slender build. The eldest brother presented with autism, mild ID, early-onset obesity and normal craniofacial features, and carried a smaller, overlapping 16p11.2 microdeletion of 847 kb (28.40–29.25 Mb), inherited from his apparently healthy father. -
Primepcr™Assay Validation Report
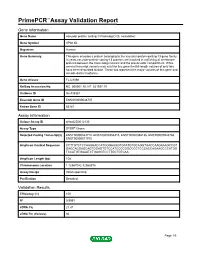
PrimePCR™Assay Validation Report Gene Information Gene Name vacuolar protein sorting 13 homolog D (S. cerevisiae) Gene Symbol VPS13D Organism Human Gene Summary This gene encodes a protein belonging to the vacuolar-protein-sorting-13 gene family. In yeast vacuolar-protein-sorting-13 proteins are involved in trafficking of membrane proteins between the trans-Golgi network and the prevacuolar compartment. While several transcript variants may exist for this gene the full-length natures of only two have been described to date. These two represent the major variants of this gene and encode distinct isoforms. Gene Aliases FLJ23066 RefSeq Accession No. NC_000001.10, NT_021937.19 UniGene ID Hs.439381 Ensembl Gene ID ENSG00000048707 Entrez Gene ID 55187 Assay Information Unique Assay ID qHsaCID0012130 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000543710, ENST00000356315, ENST00000358136, ENST00000543766, ENST00000011700 Amplicon Context Sequence CCTTGTCTCCAAAGACCATGGGAAGGTGTATGTGCAGGTGACCAAGAAAGCCGT GAGCACGAGCAGTGGAGTGTCCATCCCCGGCCCCTCCCACCAGAAGCCCATGG TCCATGTGAAATCTGAGGTCCTTGCTGTCAA Amplicon Length (bp) 108 Chromosome Location 1:12567042-12568978 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 100 R2 0.9991 cDNA Cq 21.41 cDNA Tm (Celsius) 86 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 35.2 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report VPS13D, Human Amplification Plot Amplification of -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Role of RUNX1 in Aberrant Retinal Angiogenesis Jonathan D
Page 1 of 25 Diabetes Identification of RUNX1 as a mediator of aberrant retinal angiogenesis Short Title: Role of RUNX1 in aberrant retinal angiogenesis Jonathan D. Lam,†1 Daniel J. Oh,†1 Lindsay L. Wong,1 Dhanesh Amarnani,1 Cindy Park- Windhol,1 Angie V. Sanchez,1 Jonathan Cardona-Velez,1,2 Declan McGuone,3 Anat O. Stemmer- Rachamimov,3 Dean Eliott,4 Diane R. Bielenberg,5 Tave van Zyl,4 Lishuang Shen,1 Xiaowu Gai,6 Patricia A. D’Amore*,1,7 Leo A. Kim*,1,4 Joseph F. Arboleda-Velasquez*1 Author affiliations: 1Schepens Eye Research Institute/Massachusetts Eye and Ear, Department of Ophthalmology, Harvard Medical School, 20 Staniford St., Boston, MA 02114 2Universidad Pontificia Bolivariana, Medellin, Colombia, #68- a, Cq. 1 #68305, Medellín, Antioquia, Colombia 3C.S. Kubik Laboratory for Neuropathology, Massachusetts General Hospital, 55 Fruit St., Boston, MA 02114 4Retina Service, Massachusetts Eye and Ear Infirmary, Department of Ophthalmology, Harvard Medical School, 243 Charles St., Boston, MA 02114 5Vascular Biology Program, Boston Children’s Hospital, Department of Surgery, Harvard Medical School, 300 Longwood Ave., Boston, MA 02115 6Center for Personalized Medicine, Children’s Hospital Los Angeles, Los Angeles, 4650 Sunset Blvd, Los Angeles, CA 90027, USA 7Department of Pathology, Harvard Medical School, 25 Shattuck St., Boston, MA 02115 Corresponding authors: Joseph F. Arboleda-Velasquez: [email protected] Ph: (617) 912-2517 Leo Kim: [email protected] Ph: (617) 912-2562 Patricia D’Amore: [email protected] Ph: (617) 912-2559 Fax: (617) 912-0128 20 Staniford St. Boston MA, 02114 † These authors contributed equally to this manuscript Word Count: 1905 Tables and Figures: 4 Diabetes Publish Ahead of Print, published online April 11, 2017 Diabetes Page 2 of 25 Abstract Proliferative diabetic retinopathy (PDR) is a common cause of blindness in the developed world’s working adult population, and affects those with type 1 and type 2 diabetes mellitus. -

Targeting PH Domain Proteins for Cancer Therapy
The Texas Medical Center Library DigitalCommons@TMC The University of Texas MD Anderson Cancer Center UTHealth Graduate School of The University of Texas MD Anderson Cancer Biomedical Sciences Dissertations and Theses Center UTHealth Graduate School of (Open Access) Biomedical Sciences 12-2018 Targeting PH domain proteins for cancer therapy Zhi Tan Follow this and additional works at: https://digitalcommons.library.tmc.edu/utgsbs_dissertations Part of the Bioinformatics Commons, Medicinal Chemistry and Pharmaceutics Commons, Neoplasms Commons, and the Pharmacology Commons Recommended Citation Tan, Zhi, "Targeting PH domain proteins for cancer therapy" (2018). The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access). 910. https://digitalcommons.library.tmc.edu/utgsbs_dissertations/910 This Dissertation (PhD) is brought to you for free and open access by the The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences at DigitalCommons@TMC. It has been accepted for inclusion in The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access) by an authorized administrator of DigitalCommons@TMC. For more information, please contact [email protected]. TARGETING PH DOMAIN PROTEINS FOR CANCER THERAPY by Zhi Tan Approval page APPROVED: _____________________________________________ Advisory Professor, Shuxing Zhang, Ph.D. _____________________________________________ -

Supplementary Data
Supplementary Fig. 1 A B Responder_Xenograft_ Responder_Xenograft_ NON- NON- Lu7336, Vehicle vs Lu7466, Vehicle vs Responder_Xenograft_ Responder_Xenograft_ Sagopilone, Welch- Sagopilone, Welch- Lu7187, Vehicle vs Lu7406, Vehicle vs Test: 638 Test: 600 Sagopilone, Welch- Sagopilone, Welch- Test: 468 Test: 482 Responder_Xenograft_ NON- Lu7860, Vehicle vs Responder_Xenograft_ Sagopilone, Welch - Lu7558, Vehicle vs Test: 605 Sagopilone, Welch- Test: 333 Supplementary Fig. 2 Supplementary Fig. 3 Supplementary Figure S1. Venn diagrams comparing probe sets regulated by Sagopilone treatment (10mg/kg for 24h) between individual models (Welsh Test ellipse p-value<0.001 or 5-fold change). A Sagopilone responder models, B Sagopilone non-responder models. Supplementary Figure S2. Pathway analysis of genes regulated by Sagopilone treatment in responder xenograft models 24h after Sagopilone treatment by GeneGo Metacore; the most significant pathway map representing cell cycle/spindle assembly and chromosome separation is shown, genes upregulated by Sagopilone treatment are marked with red thermometers. Supplementary Figure S3. GeneGo Metacore pathway analysis of genes differentially expressed between Sagopilone Responder and Non-Responder models displaying –log(p-Values) of most significant pathway maps. Supplementary Tables Supplementary Table 1. Response and activity in 22 non-small-cell lung cancer (NSCLC) xenograft models after treatment with Sagopilone and other cytotoxic agents commonly used in the management of NSCLC Tumor Model Response type -

Transcriptome-Wide Map of M6a Circrnas Identified in a Rat Model Of
Su et al. BMC Genomics (2020) 21:39 https://doi.org/10.1186/s12864-020-6462-y RESEARCH ARTICLE Open Access Transcriptome-wide map of m6A circRNAs identified in a rat model of hypoxia mediated pulmonary hypertension Hua Su, Guowen Wang, Lingfang Wu, Xiuqing Ma, Kejing Ying and Ruifeng Zhang* Abstract Background: Hypoxia mediated pulmonary hypertension (HPH) is a lethal disease and lacks effective therapy. CircRNAs play significant roles in physiological process. Recently, circRNAs are found to be m6A-modified. The abundance of circRNAs was influenced by m6A. Furthermore, the significance of m6A circRNAs has not been elucidated in HPH yet. Here we aim to investigate the transcriptome-wide map of m6A circRNAs in HPH. Results: Differentially expressed m6A abundance was detected in lungs of HPH rats. M6A abundance in circRNAs was significantly reduced in hypoxia in vitro. M6A circRNAs were mainly from protein-coding genes spanned single exons in control and HPH groups. Moreover, m6A influenced the circRNA–miRNA–mRNA co-expression network in hypoxia. M6A circXpo6 and m6A circTmtc3 were firstly identified to be downregulated in HPH. Conclusion: Our study firstly identified the transcriptome-wide map of m6AcircRNAsinHPH. M6A can influence circRNA–miRNA–mRNA network. Furthermore, we firstly identified two HPH-associated m6AcircRNAs: circXpo6 and circTmtc3. However, the clinical significance of m6A circRNAs for HPH should be further validated. Keywords: m6A circRNAs, Hypoxia mediated pulmonary hypertension, m6A circXpo6, m6A circTmtc3 Background Circular RNAs (circRNAs) were firstly found abundant Pulmonary hypertension (PH) is a lethal disease and defined in eukaryotes using RNA-seq approach [5–7]. Pre-mRNA as an increase in the mean pulmonary arterial pressure ≥ 25 is spliced with the 5′ and 3′ ends, forming a ‘head-to-tail’ mmHg at rest, as measured by right heart catheterization [1]. -

SUPPLEMENTARY APPENDIX Inflammation Regulates Long Non-Coding RNA-PTTG1-1:1 in Myeloid Leukemia
SUPPLEMENTARY APPENDIX Inflammation regulates long non-coding RNA-PTTG1-1:1 in myeloid leukemia Sébastien Chateauvieux, 1,2 Anthoula Gaigneaux, 1° Déborah Gérard, 1 Marion Orsini, 1 Franck Morceau, 1 Barbora Orlikova-Boyer, 1,2 Thomas Farge, 3,4 Christian Récher, 3,4,5 Jean-Emmanuel Sarry, 3,4 Mario Dicato 1 and Marc Diederich 2 °Current address: University of Luxembourg, Faculty of Science, Technology and Communication, Life Science Research Unit, Belvaux, Luxemburg. 1Laboratoire de Biologie Moléculaire et Cellulaire du Cancer, Hôpital Kirchberg, Luxembourg, Luxembourg; 2College of Pharmacy, Seoul National University, Gwanak-gu, Seoul, Korea; 3Cancer Research Center of Toulouse, UMR 1037 INSERM/ Université Toulouse III-Paul Sabatier, Toulouse, France; 4Université Toulouse III Paul Sabatier, Toulouse, France and 5Service d’Hématologie, Centre Hospitalier Universitaire de Toulouse, Institut Universitaire du Cancer de Toulouse Oncopôle, Toulouse, France Correspondence: MARC DIEDERICH - [email protected] doi:10.3324/haematol.2019.217281 Supplementary data Inflammation regulates long non-coding RNA-PTTG1-1:1 in myeloid leukemia Sébastien Chateauvieux1,2, Anthoula Gaigneaux1*, Déborah Gérard1, Marion Orsini1, Franck Morceau1, Barbora Orlikova-Boyer1,2, Thomas Farge3,4, Christian Récher3,4,5, Jean-Emmanuel Sarry3,4, Mario Dicato1 and Marc Diederich2 1 Laboratoire de Biologie Moléculaire et Cellulaire du Cancer, Hôpital Kirchberg, 9, rue Edward Steichen, 2540 Luxembourg, Luxemburg; 2 College of Pharmacy, Seoul National University, 1 Gwanak-ro,