1 Genes with 5
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
![EEF1D Mouse Monoclonal Antibody [Clone ID: OTI4B9] Product Data](https://docslib.b-cdn.net/cover/2076/eef1d-mouse-monoclonal-antibody-clone-id-oti4b9-product-data-82076.webp)
EEF1D Mouse Monoclonal Antibody [Clone ID: OTI4B9] Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for CF811676 EEF1D Mouse Monoclonal Antibody [Clone ID: OTI4B9] Product data: Product Type: Primary Antibodies Clone Name: OTI4B9 Applications: IHC, WB Recommended Dilution: WB 1:500~2000, IHC 1:2000 Reactivity: Human, Mouse, Rat Host: Mouse Isotype: IgG1 Clonality: Monoclonal Immunogen: Full length human recombinant protein of human EEF1D (NP_115754) produced in E.coli. Formulation: Lyophilized powder (original buffer 1X PBS, pH 7.3, 8% trehalose) Reconstitution Method: For reconstitution, we recommend adding 100uL distilled water to a final antibody concentration of about 1 mg/mL. To use this carrier-free antibody for conjugation experiment, we strongly recommend performing another round of desalting process. (OriGene recommends Zeba Spin Desalting Columns, 7KMWCO from Thermo Scientific) Purification: Purified from mouse ascites fluids or tissue culture supernatant by affinity chromatography (protein A/G) Conjugation: Unconjugated Storage: Store at -20°C as received. Stability: Stable for 12 months from date of receipt. Gene Name: Homo sapiens eukaryotic translation elongation factor 1 delta (EEF1D), transcript variant 1, mRNA. Database Link: NP_115754 Entrez Gene 1936 Human P29692 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 3 EEF1D Mouse Monoclonal Antibody [Clone ID: OTI4B9] – CF811676 Background: This gene encodes a subunit of the elongation factor-1 complex, which is responsible for the enzymatic delivery of aminoacyl tRNAs to the ribosome. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

A Chemical-Genetic Screen for Identifying Substrates of the Er Kinase Perk
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2014 A Chemical-Genetic Screen for Identifying Substrates of the Er Kinase Perk Nancy L. Maas University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Biology Commons, Cell Biology Commons, and the Molecular Biology Commons Recommended Citation Maas, Nancy L., "A Chemical-Genetic Screen for Identifying Substrates of the Er Kinase Perk" (2014). Publicly Accessible Penn Dissertations. 1354. https://repository.upenn.edu/edissertations/1354 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/1354 For more information, please contact [email protected]. A Chemical-Genetic Screen for Identifying Substrates of the Er Kinase Perk Abstract Cells constantly encounter changing environments that challenge the ability to adapt and survive. Signal transduction networks enable cells to appropriately sense and respond to these changes, and are often mediated through the activity of protein kinases. Protein kinases are a class of enzyme responsible for regulating a broad spectrum of cellular functions by transferring phosphate groups from ATP to substrate proteins, thereby altering substrate activity and function. PERK is a resident kinase of the endoplasmic reticulum, and is responsible for sensing perturbations in the protein folding capacity of the ER. When the influx of unfolded, nascent proteins exceeds the folding capacity of the ER, PERK initiates a cascade of signaling events that enable cell adaptation and ER stress resolution. These signaling pathways are not only essential for the survival of normal cells undergoing ER stress, but are also co-opted by tumor cells in order to survive the oxygen and nutrient-restricted conditions of the tumor microenvironment. -

Genes with 5' Terminal Oligopyrimidine Tracts Preferentially Escape Global Suppression of Translation by the SARS-Cov-2 NSP1 Protein
Downloaded from rnajournal.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press Genes with 5′ terminal oligopyrimidine tracts preferentially escape global suppression of translation by the SARS-CoV-2 Nsp1 protein Shilpa Raoa, Ian Hoskinsa, Tori Tonna, P. Daniela Garciaa, Hakan Ozadama, Elif Sarinay Cenika, Can Cenika,1 a Department of Molecular Biosciences, University of Texas at Austin, Austin, TX 78712, USA 1Corresponding author: [email protected] Key words: SARS-CoV-2, Nsp1, MeTAFlow, translation, ribosome profiling, RNA-Seq, 5′ TOP, Ribo-Seq, gene expression 1 Downloaded from rnajournal.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract Viruses rely on the host translation machinery to synthesize their own proteins. Consequently, they have evolved varied mechanisms to co-opt host translation for their survival. SARS-CoV-2 relies on a non-structural protein, Nsp1, for shutting down host translation. However, it is currently unknown how viral proteins and host factors critical for viral replication can escape a global shutdown of host translation. Here, using a novel FACS-based assay called MeTAFlow, we report a dose-dependent reduction in both nascent protein synthesis and mRNA abundance in cells expressing Nsp1. We perform RNA-Seq and matched ribosome profiling experiments to identify gene-specific changes both at the mRNA expression and translation level. We discover that a functionally-coherent subset of human genes are preferentially translated in the context of Nsp1 expression. These genes include the translation machinery components, RNA binding proteins, and others important for viral pathogenicity. Importantly, we uncovered a remarkable enrichment of 5′ terminal oligo-pyrimidine (TOP) tracts among preferentially translated genes. -

Apoptotic Genes As Potential Markers of Metastatic Phenotype in Human Osteosarcoma Cell Lines
17-31 10/12/07 14:53 Page 17 INTERNATIONAL JOURNAL OF ONCOLOGY 32: 17-31, 2008 17 Apoptotic genes as potential markers of metastatic phenotype in human osteosarcoma cell lines CINZIA ZUCCHINI1, ANNA ROCCHI2, MARIA CRISTINA MANARA2, PAOLA DE SANCTIS1, CRISTINA CAPANNI3, MICHELE BIANCHINI1, PAOLO CARINCI1, KATIA SCOTLANDI2 and LUISA VALVASSORI1 1Dipartimento di Istologia, Embriologia e Biologia Applicata, Università di Bologna, Via Belmeloro 8, 40126 Bologna; 2Laboratorio di Ricerca Oncologica, Istituti Ortopedici Rizzoli; 3IGM-CNR, Unit of Bologna, c/o Istituti Ortopedici Rizzoli, Via di Barbiano 1/10, 40136 Bologna, Italy Received May 29, 2007; Accepted July 19, 2007 Abstract. Metastasis is the most frequent cause of death among malignant primitive bone tumor, usually developing in children patients with osteosarcoma. We have previously demonstrated and adolescents, with a high tendency to metastasize (2). in independent experiments that the forced expression of Metastases in osteosarcoma patients spread through peripheral L/B/K ALP and CD99 in U-2 OS osteosarcoma cell lines blood very early and colonize primarily the lung, and later markedly reduces the metastatic ability of these cancer cells. other skeleton districts (3). Since disseminated hidden micro- This behavior makes these cell lines a useful model to assess metastases are present in 80-90% of OS patients at the time the intersection of multiple and independent gene expression of diagnosis, the identification of markers of invasiveness signatures concerning the biological problem of dissemination. and metastasis forms a target of paramount importance in With the aim to characterize a common transcriptional profile planning the treatment of osteosarcoma lesions and enhancing reflecting the essential features of metastatic behavior, we the prognosis. -

Role of Cyclin-Dependent Kinase 1 in Translational Regulation in the M-Phase
cells Review Role of Cyclin-Dependent Kinase 1 in Translational Regulation in the M-Phase Jaroslav Kalous *, Denisa Jansová and Andrej Šušor Institute of Animal Physiology and Genetics, Academy of Sciences of the Czech Republic, Rumburska 89, 27721 Libechov, Czech Republic; [email protected] (D.J.); [email protected] (A.Š.) * Correspondence: [email protected] Received: 28 April 2020; Accepted: 24 June 2020; Published: 27 June 2020 Abstract: Cyclin dependent kinase 1 (CDK1) has been primarily identified as a key cell cycle regulator in both mitosis and meiosis. Recently, an extramitotic function of CDK1 emerged when evidence was found that CDK1 is involved in many cellular events that are essential for cell proliferation and survival. In this review we summarize the involvement of CDK1 in the initiation and elongation steps of protein synthesis in the cell. During its activation, CDK1 influences the initiation of protein synthesis, promotes the activity of specific translational initiation factors and affects the functioning of a subset of elongation factors. Our review provides insights into gene expression regulation during the transcriptionally silent M-phase and describes quantitative and qualitative translational changes based on the extramitotic role of the cell cycle master regulator CDK1 to optimize temporal synthesis of proteins to sustain the division-related processes: mitosis and cytokinesis. Keywords: CDK1; 4E-BP1; mTOR; mRNA; translation; M-phase 1. Introduction 1.1. Cyclin Dependent Kinase 1 (CDK1) Is a Subunit of the M Phase-Promoting Factor (MPF) CDK1, a serine/threonine kinase, is a catalytic subunit of the M phase-promoting factor (MPF) complex which is essential for cell cycle control during the G1-S and G2-M phase transitions of eukaryotic cells. -
Type of the Paper (Article
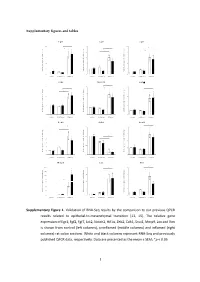
Supplementary figures and tables E g r 1 F g f2 F g f7 1 0 * 5 1 0 * * e e e * g g g * n n n * a a a 8 4 * 8 h h h * c c c d d d * l l l o o o * f f f * n n n o o o 6 3 6 i i i s s s s s s e e e r r r p p p x x x e e e 4 2 4 e e e n n n e e e g g g e e e v v v i i i t t t 2 1 2 a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d J a k 2 N o tc h 2 H if1 * 3 4 6 * * * e e e g g g n n n a a * * a * h h * h c c c 3 * d d * d l l l * o o o f f 2 f 4 n n n o o o i i i s s s s s s e e e r r 2 r p p p x x x e e e e e e n n n e e 1 e 2 g g g e e 1 e v v v i i i t t t a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Z e b 2 C d h 1 S n a i1 * * 7 1 .5 4 * * e e e g g g 6 n n n * a a a * h h h c c c 3 * d d d l l l 5 o o o f f f 1 .0 * n n n * o o o i i i 4 * s s s s s s e e e r r r 2 p p p x x x 3 e e e e e e n n n e e e 0 .5 g g g 2 e e e 1 v v v i i i t t t a a a * l l l e e e 1 * R R R 0 0 .0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d M m p 9 L o x V im 2 0 0 2 0 8 * * * e e e * g g g 1 5 0 * n n n * a a a * h h h * c c c 1 5 * 6 d d d l l l 1 0 0 o o o f f f n n n o o o i i i 5 0 s s s s s s * e e e r r r 1 0 4 3 0 p p p * x x x e e e * e e e n n n e e e 2 0 g g g e e e 5 2 v v v i i i t t t a a a l l l 1 0 e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Supplementary Figure 1. -

Global Analysis of LARP1 Translation Targets Reveals Tunable and Dynamic Features of 5′ TOP Motifs
Global analysis of LARP1 translation targets reveals tunable and dynamic features of 5′ TOP motifs Lucas Philippea,1, Antonia M. G. van den Elzena,1, Maegan J. Watsona, and Carson C. Thoreena,2 aDepartment of Cellular and Molecular Physiology, Yale School of Medicine, New Haven, CT 06510 Edited by Alan G. Hinnebusch, National Institutes of Health, Bethesda, MD, and approved January 29, 2020 (received for review July 25, 2019) Terminal oligopyrimidine (TOP) motifs are sequences at the 5′ ends recent findings have hinted that the RNA-binding protein La- of mRNAs that link their translation to the mTOR Complex 1 related protein 1 (LARP1) may have a central role (8–10). (mTORC1) nutrient-sensing signaling pathway. They are com- LARP1 is a large protein (150 kDa) with several RNA-binding monly regarded as discrete elements that reside on ∼100 mRNAs domains. Its central region contains a La motif (LaM) domain that mostly encode translation factors. However, the full spectrum that defines the La-related protein (LARP) superfamily, along of TOP sequences and their prevalence throughout the transcrip- with an adjacent RNA recognition motif-like (RRM-L) domain. tome remain unclear, primarily because of uncertainty over the Its C terminus encodes a domain known as the DM15 region. This mechanism that detects them. Here, we globally analyzed trans- domain is unique to LARP1 and its closely related homolog lation targets of La-related protein 1 (LARP1), an RNA-binding pro- LARP1B, and is therefore also known as the LARP1 domain (11). tein and mTORC1 effector that has been shown to repress TOP Several observations suggest that LARP1 directly represses TOP mRNA translation in a few specific cases. -

Estrogenic Endocrine Disrupting Chemicals Influencing NRF1 Regulated Gene Networks in the Development of Complex Human Brain
Florida International University FIU Digital Commons All Faculty 12-13-2016 Estrogenic Endocrine Disrupting Chemicals Influencing NRF1 Regulated Gene Networks in the Development of Complex Human Brain Diseases Mark Preciados Department of Environmental & Occupational Health, Florida International University, [email protected] Changwon Yoo Department of Biostatistics, Florida International University, [email protected] Deodutta Roy Department of Environmental & Occupational Health, Florida International University, [email protected] Follow this and additional works at: https://digitalcommons.fiu.edu/all_faculty Recommended Citation Preciados, Mark; Yoo, Changwon; and Roy, Deodutta, "Estrogenic Endocrine Disrupting Chemicals Influencing NRF1 Regulated Gene Networks in the Development of Complex Human Brain Diseases" (2016). All Faculty. 180. https://digitalcommons.fiu.edu/all_faculty/180 This work is brought to you for free and open access by FIU Digital Commons. It has been accepted for inclusion in All Faculty by an authorized administrator of FIU Digital Commons. For more information, please contact [email protected]. International Journal of Molecular Sciences Review Estrogenic Endocrine Disrupting Chemicals Influencing NRF1 Regulated Gene Networks in the Development of Complex Human Brain Diseases Mark Preciados 1, Changwon Yoo 2 and Deodutta Roy 1,* 1 Department of Environmental & Occupational Health, Florida International University, Miami, FL 33199, USA; mprec001@fiu.edu 2 Department of Biostatistics, Florida International University, Miami, FL 33199, USA; cyoo@fiu.edu * Correspondence: droy@fiu.edu; Tel.: +1-305-348-1694; Fax: +1-305-348-4901 Academic Editor: Paul B. Tchounwou Received: 25 October 2016; Accepted: 29 November 2016; Published: 13 December 2016 Abstract: During the development of an individual from a single cell to prenatal stages to adolescence to adulthood and through the complete life span, humans are exposed to countless environmental and stochastic factors, including estrogenic endocrine disrupting chemicals. -

New Partners Identified by Mass Spectrometry Assay Reveal Functions of NCAM2 in Neural Cytoskeleton Organization
International Journal of Molecular Sciences Article New Partners Identified by Mass Spectrometry Assay Reveal Functions of NCAM2 in Neural Cytoskeleton Organization Antoni Parcerisas 1,2,3,*,† , Alba Ortega-Gascó 1,2,† , Marc Hernaiz-Llorens 1,2 , Maria Antonia Odena 4, Fausto Ulloa 1,2, Eliandre de Oliveira 4, Miquel Bosch 3 , Lluís Pujadas 1,2 and Eduardo Soriano 1,2,* 1 Department of Cell Biology, Physiology and Immunology, University of Barcelona and Institute of Neurosciences, 08028 Barcelona, Spain; [email protected] (A.O.-G.); [email protected] (M.H.-L.); [email protected] (F.U.); [email protected] (L.P.) 2 Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas (CIBERNED), 28031 Madrid, Spain 3 Department of Basic Sciences, Universitat Internacional de Catalunya, 08195 Sant Cugat del Vallès, Spain; [email protected] 4 Plataforma de Proteòmica, Parc Científic de Barcelona (PCB), 08028 Barcelona, Spain; [email protected] (M.A.O.); [email protected] (E.d.O.) * Correspondence: [email protected] (A.P.); [email protected] (E.S.) † A.P. and A.O.-G. contributed equally. Abstract: Neuronal cell adhesion molecule 2 (NCAM2) is a membrane protein with an important role in the morphological development of neurons. In the cortex and the hippocampus, NCAM2 is essential for proper neuronal differentiation, dendritic and axonal outgrowth and synapse forma- tion. However, little is known about NCAM2 functional mechanisms and its interactive partners Citation: Parcerisas, A.; during brain development. Here we used mass spectrometry to study the molecular interactome Ortega-Gascó, A.; Hernaiz-Llorens, of NCAM2 in the second postnatal week of the mouse cerebral cortex. -

Overview of Research on Fusion Genes in Prostate Cancer
2011 Review Article Overview of research on fusion genes in prostate cancer Chunjiao Song1,2, Huan Chen3 1Medical Research Center, Shaoxing People’s Hospital, Shaoxing University School of Medicine, Shaoxing 312000, China; 2Shaoxing Hospital, Zhejiang University School of Medicine, Shaoxing 312000, China; 3Key Laboratory of Microorganism Technology and Bioinformatics Research of Zhejiang Province, Zhejiang Institute of Microbiology, Hangzhou 310000, China Contributions: (I) Conception and design: C Song; (II) Administrative support: Shaoxing Municipal Health and Family Planning Science and Technology Innovation Project (2017CX004) and Shaoxing Public Welfare Applied Research Project (2018C30058); (III) Provision of study materials or patients: None; (IV) Collection and assembly of data: C Song; (V) Data analysis and interpretation: H Chen; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. Correspondence to: Chunjiao Song. No. 568 Zhongxing Bei Road, Shaoxing 312000, China. Email: [email protected]. Abstract: Fusion genes are known to drive and promote carcinogenesis and cancer progression. In recent years, the rapid development of biotechnologies has led to the discovery of a large number of fusion genes in prostate cancer specimens. To further investigate them, we summarized the fusion genes. We searched related articles in PubMed, CNKI (Chinese National Knowledge Infrastructure) and other databases, and the data of 92 literatures were summarized after preliminary screening. In this review, we summarized approximated 400 fusion genes since the first specific fusion TMPRSS2-ERG was discovered in prostate cancer in 2005. Some of these are prostate cancer specific, some are high-frequency in the prostate cancer of a certain ethnic group. This is a summary of scientific research in related fields and suggests that some fusion genes may become biomarkers or the targets for individualized therapies. -

Diagnostic Yield and Novel Candidate Genes by Exome Sequencing in 152 Consanguineous Families with Neurodevelopmental Disorders
Research JAMA Psychiatry | Original Investigation Diagnostic Yield and Novel Candidate Genes by Exome Sequencing in 152 Consanguineous Families With Neurodevelopmental Disorders Miriam S. Reuter, MD; Hasan Tawamie, MA; Rebecca Buchert, MA; Ola Hosny Gebril, MD; Tawfiq Froukh, PhD; Christian Thiel, MD; Steffen Uebe, PhD; Arif B. Ekici, PhD; Mandy Krumbiegel, PhD; Christiane Zweier, MD; Juliane Hoyer, MD; Karolin Eberlein, MD; Judith Bauer, MD; Ute Scheller, MD; Tim M. Strom, MD; Sabine Hoffjan, MD; Ehab R. Abdelraouf, MD; Nagwa A. Meguid, MD, PhD; Ahmad Abboud, MD; Mohammed Ayman Al Khateeb, MD; Mahmoud Fakher, MD; Saber Hamdan, MD; Amina Ismael, MD; Safia Muhammad, MD; Ebtessam Abdallah, MD, PhD; Heinrich Sticht, PhD; Dagmar Wieczorek, MD; André Reis, MD; Rami Abou Jamra, MD Supplemental content IMPORTANCE Autosomal recessive inherited neurodevelopmental disorders are highly heterogeneous, and many, possibly most, of the disease genes are still unknown. OBJECTIVES To promote the identification of disease genes through confirmation of previously described genes and presentation of novel candidates and provide an overview of the diagnostic yield of exome sequencing in consanguineous families. DESIGN, SETTING, AND PARTICIPANTS Autozygosity mapping in families and exome sequencing of index patients were performed in 152 consanguineous families (the parents descended from a same ancestor) with at least 1 offspring with intellectual disability (ID). The study was conducted from July 1, 2008, to June 30, 2015, and data analysis was conducted from July 1, 2015, to August 31, 2016. RESULTS Of the 152 consanguineous families enrolled, 1 child (in 45 families [29.6%]) or multiple children (107 families [70.4%]) had ID; additional features were present in 140 of the families (92.1%).