TELOMERE-ASSOCIATED PROTEINS in Arabidopsis Thaliana
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Open-Sourced Bioinformatic Pipeline for the Processing of Next-Generation Sequencing Derived Nucleotide Reads
bioRxiv preprint doi: https://doi.org/10.1101/2020.04.20.050369; this version posted May 28, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. An open-sourced bioinformatic pipeline for the processing of Next-Generation Sequencing derived nucleotide reads: Identification and authentication of ancient metagenomic DNA Thomas C. Collin1, *, Konstantina Drosou2, 3, Jeremiah Daniel O’Riordan4, Tengiz Meshveliani5, Ron Pinhasi6, and Robin N. M. Feeney1 1School of Medicine, University College Dublin, Ireland 2Division of Cell Matrix Biology Regenerative Medicine, University of Manchester, United Kingdom 3Manchester Institute of Biotechnology, School of Earth and Environmental Sciences, University of Manchester, United Kingdom [email protected] 5Institute of Paleobiology and Paleoanthropology, National Museum of Georgia, Tbilisi, Georgia 6Department of Evolutionary Anthropology, University of Vienna, Austria *Corresponding Author Abstract The emerging field of ancient metagenomics adds to these Bioinformatic pipelines optimised for the processing and as- processing complexities with the need for additional steps sessment of metagenomic ancient DNA (aDNA) are needed in the separation and authentication of ancient sequences from modern sequences. Currently, there are few pipelines for studies that do not make use of high yielding DNA cap- available for the analysis of ancient metagenomic DNA ture techniques. These bioinformatic pipelines are tradition- 1 4 ally optimised for broad aDNA purposes, are contingent on (aDNA) ≠ The limited number of bioinformatic pipelines selection biases and are associated with high costs. -

Identification of Transcribed Sequences in Arabidopsis Thaliana by Using High-Resolution Genome Tiling Arrays
Identification of transcribed sequences in Arabidopsis thaliana by using high-resolution genome tiling arrays Viktor Stolc*†‡§, Manoj Pratim Samanta‡§¶, Waraporn Tongprasitʈ, Himanshu Sethiʈ, Shoudan Liang*, David C. Nelson**, Adrian Hegeman**, Clark Nelson**, David Rancour**, Sebastian Bednarek**, Eldon L. Ulrich**, Qin Zhao**, Russell L. Wrobel**, Craig S. Newman**, Brian G. Fox**, George N. Phillips, Jr.**, John L. Markley**, and Michael R. Sussman**†† *Genome Research Facility, National Aeronautics and Space Administration Ames Research Center, Moffett Field, CA 94035; †Department of Molecular, Cellular, and Developmental Biology, Yale University, New Haven, CT 06520; ¶Systemix Institute, Cupertino, CA 94035; ʈEloret Corporation at National Aeronautics and Space Administration Ames Research Center, Moffett Field, CA 94035; and **Center for Eukaryotic Structural Genomics, University of Wisconsin, Madison, WI 53706 Edited by Sidney Altman, Yale University, New Haven, CT, and approved January 28, 2005 (received for review November 4, 2004) Using a maskless photolithography method, we produced DNA Genome-wide tiling arrays can overcome many of the shortcom- oligonucleotide microarrays with probe sequences tiled through- ings of the previous approaches by comprehensively probing out the genome of the plant Arabidopsis thaliana. RNA expression transcription in all regions of the genome. This technology has was determined for the complete nuclear, mitochondrial, and been used successfully on different organisms (5–12). A recent chloroplast genomes by tiling 5 million 36-mer probes. These study on A. thaliana reported measuring transcriptional activities probes were hybridized to labeled mRNA isolated from liquid of four different cell lines by using 25-mer-based tiling arrays that grown T87 cells, an undifferentiated Arabidopsis cell culture line. -

Efficient Storage and Analysis of Genome Data in Relational Database Systems
Efficient Storage and Analysis of Genome Data in Relational Database Systems D I S S E R T A T I O N zur Erlangung des akademischen Grades Doktoringenieur (Dr.-Ing.) angenommen durch die Fakultät für Informatik der Otto-von-Guericke-Universität Magdeburg von M.Sc. Sebastian Dorok geb. am 09.10.1986 in Haldensleben Gutachterinnen/Gutachter Prof. Dr. Gunter Saake Prof. Dr. Jens Teubner Prof. Dr. Ralf Hofestädt Magdeburg, den 27.04.2017 Dorok, Sebastian: Efficient storage and analysis of genome data in relational database systems Dissertation, University of Magdeburg, 2017. Abstract Genome analysis allows researchers to reveal insights about the genetic makeup of living organisms. In the near future, genome analysis will become a key means in the detection and treatment of diseases that are based on variations of the genetic makeup. To this end, powerful variant detection tools were developed or are still under development. However, genome analysis faces a large data deluge. The amounts of data that are produced in a typical genome analysis experiment easily exceed several 100 gigabytes. At the same time, the number of genome analysis experiments increases as the costs drop. Thus, the reliable and efficient management and analysis of large amounts of genome data will likely become a bottleneck, if we do not improve current genome data management and analysis solutions. Currently, genome data management and analysis relies mainly on flat-file based storage and command-line driven analysis tools. Such approaches offer only limited data man- agement capabilities that can hardly cope with future requirements such as annotation management or provenance tracking. -

Annual Scientific Report 2011 Annual Scientific Report 2011 Designed and Produced by Pickeringhutchins Ltd
European Bioinformatics Institute EMBL-EBI Annual Scientific Report 2011 Annual Scientific Report 2011 Designed and Produced by PickeringHutchins Ltd www.pickeringhutchins.com EMBL member states: Austria, Croatia, Denmark, Finland, France, Germany, Greece, Iceland, Ireland, Israel, Italy, Luxembourg, the Netherlands, Norway, Portugal, Spain, Sweden, Switzerland, United Kingdom. Associate member state: Australia EMBL-EBI is a part of the European Molecular Biology Laboratory (EMBL) EMBL-EBI EMBL-EBI EMBL-EBI EMBL-European Bioinformatics Institute Wellcome Trust Genome Campus, Hinxton Cambridge CB10 1SD United Kingdom Tel. +44 (0)1223 494 444, Fax +44 (0)1223 494 468 www.ebi.ac.uk EMBL Heidelberg Meyerhofstraße 1 69117 Heidelberg Germany Tel. +49 (0)6221 3870, Fax +49 (0)6221 387 8306 www.embl.org [email protected] EMBL Grenoble 6, rue Jules Horowitz, BP181 38042 Grenoble, Cedex 9 France Tel. +33 (0)476 20 7269, Fax +33 (0)476 20 2199 EMBL Hamburg c/o DESY Notkestraße 85 22603 Hamburg Germany Tel. +49 (0)4089 902 110, Fax +49 (0)4089 902 149 EMBL Monterotondo Adriano Buzzati-Traverso Campus Via Ramarini, 32 00015 Monterotondo (Rome) Italy Tel. +39 (0)6900 91402, Fax +39 (0)6900 91406 © 2012 EMBL-European Bioinformatics Institute All texts written by EBI-EMBL Group and Team Leaders. This publication was produced by the EBI’s Outreach and Training Programme. Contents Introduction Foreword 2 Major Achievements 2011 4 Services Rolf Apweiler and Ewan Birney: Protein and nucleotide data 10 Guy Cochrane: The European Nucleotide Archive 14 Paul Flicek: -

CRAM Format Specification (Version 3.0)
CRAM format specification (version 3.0) [email protected] 4 Feb 2021 The master version of this document can be found at https://github.com/samtools/hts-specs. This printing is version c467e20 from that repository, last modified on the date shown above. license: Apache 2.0 1 Overview This specification describes the CRAM 3.0 format. CRAM has the following major objectives: 1. Significantly better lossless compression than BAM 2. Full compatibility with BAM 3. Effortless transition to CRAM from using BAM files 4. Support for controlled loss of BAM data The first three objectives allow users to take immediate advantage of the CRAM format while offering a smooth transition path from using BAM files. The fourth objective supports the exploration of different lossy compres- sion strategies and provides a framework in which to effect these choices. Please note that the CRAM format does not impose any rules about what data should or should not be preserved. Instead, CRAM supports a wide range of lossless and lossy data preservation strategies enabling users to choose which data should be preserved. Data in CRAM is stored either as CRAM records or using one of the general purpose compressors (gzip, bzip2). CRAM records are compressed using a number of different encoding strategies. For example, bases are reference compressed by encoding base differences rather than storing the bases themselves.1 2 Data types CRAM specification uses logical data types and storage data types; logical data types are written as words (e.g. int) while physical data types are written using single letters (e.g. -

Htslib - C Library for Reading/Writing High-Throughput Sequencing Data
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.16.423064; this version posted December 16, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. HTSlib - C library for reading/writing high-throughput sequencing data Authors: James K. Bonfield*, John Marshall*, Petr Danecek*, Heng Li, Valeriu Ohan, Andrew Whitwham, Thomas Keane, Robert M. Davies (* contributed equally) Abstract Background Since the original publication of the VCF and SAM formats, an explosion of software tools have been created to process these data files. To facilitate this a library was produced out of the original SAMtools implementation, with a focus on performance and robustness. The file formats themselves have become international standards under the jurisdiction of the Global Alliance for Genomics and Health. Findings We present a software library for providing programmatic access to sequencing alignment and variant formats. It was born out of the widely used SAMtools and BCFtools applications. Considerable improvements have been made to the original code plus many new features including newer access protocols, the addition of the CRAM file format, better indexing and iterators, and better use of threading. Conclusion Since the original Samtools release, performance has been considerably improved, with a BAM read-write loop running 4.9 times faster and BAM to SAM conversion at 12.8 times faster (both using 16 threads, compared to Samtools 0.1.19). Widespread adoption has seen HTSlib downloaded over a million times from GitHub and conda. -

Which Genome Browser to Use for My Data ? July 2019
Which genome browser to use for my data ? July 2019 The development of genome sequencing projects since the early 2000s has been accompanied by efforts from the scientific community to develop interactive graphical visualisation tools, called genome browsers or genome viewers. This effort has been further intensified with the advent of high throughput sequencing and the need to visualise data as diverse as Whole Genome Sequencing, exomes, RNA-seq, ChIP-seq, variants, interactions, in connection with publicly available annotation information. Over the years, genome viewers have become increasingly sophisticated tools with advanced features for data exploration and interpretation. We have reviewed seven genome browsers, selected (arbitrarily) for their notoriety and their complementarity: Artemis, GIVE, IGB, IGV, Jbrowse, Tablet, UCSC Genome Browser. The purpose of this study is to provide simple guidelines to help you to choose the right genome browser software for your next project. Our selection is of course far from being exhaustive. There are many other tools of interest that could have been included: Genomeview, Ensembl genome browser, Savant for example. We noted also the existence of many specialized viewers: WashU for epigenetics, Ribbon for third generation sequencing, (3rd gen), Single cell genome viewer, Asciigenome,... If you are interested in this work and would like to contribute it, feel free to contact us: [email protected] We invite you to take a tour. 1. Identity forms 2. Methodology, datasets 3. Technical features 4. Ease of use 5. Supported types of data 6. Navigation, visualisation 7. RAM and time requirements 8. Sharing facilities, interoperability 9. A few words of conclusion 10. -

Training Modules Documentation Release 1
training_modules Documentation Release 1 Marc B Rossello Sep 24, 2021 ENA Data Submission 1 General Guide on ENA Data Submission3 2 How to Register a Study 29 3 How to Register Samples 35 4 Preparing Files for Submission 47 5 How to Submit Raw Reads 83 6 How to Submit Assemblies 103 7 How to Submit Targeted Sequences 145 8 How to Submit Other Analyses 161 9 General Guide on ENA Data Retrieval 183 10 Updating Metadata Objects 223 11 Updating Assemblies 231 12 Updating Annotated Sequences 233 13 Data Release Policies 235 14 Common Run Submission Errors 237 15 Common Sample Submission Errors 243 16 Tips for Sample Taxonomy 245 17 Requesting New Taxon IDs 249 18 Metagenome Submission Queries 255 19 Locus Tag Prefixes 261 20 Archive Generated FASTQ Files 263 i 21 Third Party Tools 269 22 Introductory Webinar 271 ii training_modules Documentation, Release 1 Welcome to the guidelines for submission and retrieval for the European Nucleotide Archive. Please use the links to find instructions specific to your needs. If you’re completely new to ENA, you can see an introductory webinar at the bottom of the page. ENA Data Submission 1 training_modules Documentation, Release 1 2 ENA Data Submission CHAPTER 1 General Guide on ENA Data Submission Welcome to the general guide for the European Nucleotide Archive submission. Please take a moment to view this introduction and consider the options available to you before you begin your submission. ENA allows submissions via three routes, each of which is appropriate for a different set of submission types. -

Three Data Delivery Cases for EMBL- EBI's Embassy
Three data delivery cases for EMBL- EBI’s Embassy Guy Cochrane www.ebi.ac.uk EMBL European Bioinformatics Institute Genes, genomes & variation Protein sequences • European Nucleotide Archive • InterPro • 1000 Genomes • Pfam • Ensembl • UniProt • Ensembl Genomes • Ensembl Plants Molecular structures • European Genome-phenome Archive • Protein Data Bank in Europe • Metagenomics portal • Electron Microscopy Data Bank • GWAS Catalog browser Expression • ArrayExpress Chemical biology • Expression Atlas • ChEMBL • Metabolights • ChEBI • PRIDE Literature & ontology • Europe PubMed Central Reactions, interactions • Gene Ontology & pathways Systems • Experimental Factor • IntAct • BioModels Ontology • Reactome • Enzyme Portal • MetaboLights • BioSamples Sequence data at EMBL-EBI Sample/method Sample/method Read Read Alignment Alignment European Genome-phenome Archive - Controlled access data - Human data around molecular medicine Assembly - http://www.ebi.ac.uk/ega/ Annotation European Nucleotide Archive - Unrestricted data - Pan-species and application - http://www.ebi.ac.uk/ena/ Sequence data at EMBL-EBI Sample/method Sample/method Read Read Alignment Alignment European Genome-phenome Archive - Controlled access data - Human data around molecular medicine Assembly - http://www.ebi.ac.uk/ega/ Infrastructure provision Annotation - BBSRC: RNAcentral, MG Portal - MRC: 100k Genomes data implementation European Nucleotide Archive - EC: COMPARE, MicroB3, ESGI, - Unrestricted data BASIS - Pan-species and application - http://www.ebi.ac.uk/ena/ - etc. -

An Integrated Pipeline for Metagenomic Taxonomy Identification and Compression Minji Kim1* , Xiejia Zhang1,Jonathang.Ligo1, Farzad Farnoud2, Venugopal V
Kim et al. BMC Bioinformatics (2016) 17:94 DOI 10.1186/s12859-016-0932-x METHODOLOGY ARTICLE Open Access MetaCRAM: an integrated pipeline for metagenomic taxonomy identification and compression Minji Kim1* , Xiejia Zhang1,JonathanG.Ligo1, Farzad Farnoud2, Venugopal V. Veeravalli1 and Olgica Milenkovic1 Abstract Background: Metagenomics is a genomics research discipline devoted to the study of microbial communities in environmental samples and human and animal organs and tissues. Sequenced metagenomic samples usually comprise reads from a large number of different bacterial communities and hence tend to result in large file sizes, typically ranging between 1–10 GB. This leads to challenges in analyzing, transferring and storing metagenomic data. In order to overcome these data processing issues, we introduce MetaCRAM, the first de novo, parallelized software suite specialized for FASTA and FASTQ format metagenomic read processing and lossless compression. Results: MetaCRAM integrates algorithms for taxonomy identification and assembly, and introduces parallel execution methods; furthermore, it enables genome reference selection and CRAM based compression. MetaCRAM also uses novel reference-based compression methods designed through extensive studies of integer compression techniques and through fitting of empirical distributions of metagenomic read-reference positions. MetaCRAM is a lossless method compatible with standard CRAM formats, and it allows for fast selection of relevant files in the compressed domain via maintenance of taxonomy information. The performance of MetaCRAM as a stand-alone compression platform was evaluated on various metagenomic samples from the NCBI Sequence Read Archive, suggesting 2- to 4-fold compression ratio improvements compared to gzip. On average, the compressed file sizes were 2-13 percent of the original raw metagenomic file sizes. -
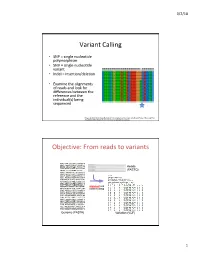
Variant Calling Objective: from Reads to Variants
3/2/18 Variant Calling • SNP = single nucleotide polymorphism • SNV = single nucleotide variant • Indel = insertion/deletion • Examine the alignments of reads and look for differences between the reference and the individual(s) being sequenced Feng et al 2014. Novel Single Nucleotide Polymorphisms of the Insulin-Like Growth Factor-I Gene and Their Associations with Growth Traits in Common Carp (Cyprinus carpio L.) Objective: From reads to variants Reads (FASTQ) alignment and variant calling Genome (FASTA) Variation (VCF) 1 3/2/18 Your mileage may vary • Different decisions about how to align reads and identify variants can yield very different results • 5 pipelines • “SNP concordance between five Illumina pipelines across all 15 exomes was 57.4%, while 0.5 to 5.1% of variants were called as unique to each pipeline. Indel concordance was only 26.8% between three indel-calling pipelines” Variant Calling Difficulties • Difficulties: – Cloning process (PCR) artifacts – Errors in the sequencing reads – Incorrect mapping – Errors in the reference genome • Heng Li, developer of BWA, looked at major sources of errors in variant calls*: – erroneous realignment in low-complexity (tandem) regions – the incomplete reference genome with respect to the sample * Li 2014 Toward better understanding of artifacts in variant calling from high- coverage samples. Bioinformatics. 2 3/2/18 Indel Can be difficult to decide where the best alignment actually is. Indels are far more problematic to call than SNPs. *https://bioinf.comav.upv.es/courses/sequence_analysis/snp_calling.html -

Future Development of the Samtools Software Package John Marshall, Petr Daněček, James K
Future development of the Samtools software package John Marshall, Petr Daněček, James K. Bonfield, Robert M. Davies, Martin O. Pollard, Shane A. McCarthy,# Thomas M. Keane, Heng Li, Richard Durbin Vertebrate Resequencing, Wellcome Trust Sanger Institute, Hinxton, United Kingdom Samtools is one of the most widely-used software packages for HTSLIB analysing next-generation sequencing data. Since the release# C library for handling high-throughput sequencing data, providing APIs for manipulating SAM, BAM, and CRAM & reference-based compression of version 0.1.19 in March 2013, there have been over forty CRAM sequence files (similar to but more flexible than the old Samtools API) and for manipulating VCF or CRAM is a format developed by the European Bioinformatics thousand downloads of Samtools. Samtools and its associated BCF variant files. Used by samtools and bcftools and also by other programmers in their C programs. Institute to exploit higher compression gained by reordering data sub-tools are used throughout many NGS pipelines for data and by the use of a known reference sequence. processing tasks such as creating, converting, indexing, viewing, SAMTOOLS BCFTOOLS Sequence names look like other names and quality values look like sorting, and merging SAM, BAM, VCF, and BCF files. Bcftools, Tools for manipulating SAM, BAM, and CRAM Tools for manipulating VCF and BCF files, and for other quality values, so compress well individually; however mixing also part of the package, is used for SNP and indel calling and alignment files, and for producing pileups and BCF variant calling, notably: the two data types leads to more disparity and less compression.