Thesis for Word XP
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Girl Names Registered in 1996
Baby Girl Names Registered in 1996 # Baby Girl Names # Baby Girl Names # Baby Girl Names 1 Aaliyah 1 Aiesha 1 Aleeta 1 Aamino 2 Aileen 1 Aleigha 1 Aamna 1 Ailish 2 Aleksandra 1 Aanchal 1 Ailsa 3 Alena 2 Aaryn 4 Aimee 1 Alesha 1 Aashna 1Ainslay 1 Alesia 5 Abbey 1Ainsleigh 1 Alesian 1 Abbi 4Ainsley 6 Alessandra 3 Abbie 1 Airianna 1 Alessia 2 Abbigail 1Airyn 1 Aleta 19 Abby 4 Aisha 5 Alex 1 Abear 1 Aishling 25 Alexa 1 Abena 6 Aislinn 1 Alexander 1 Abigael 1 Aiyana-Marie 128 Alexandra 32 Abigail 2Aja 2 Alexandrea 5 Abigayle 1 Ajdina 29 Alexandria 2 Abir 1 Ajsha 5 Alexia 1 Abrianna 1 Akasha 49 Alexis 1 Abrinna 1Akayla 1 Alexsandra 1 Abyen 2Akaysha 1 Alexus 1 Abygail 1Akelyn 2 Ali 2 Acacia 1 Akosua 7 Alia 1 Accacca 1 Aksana 1 Aliah 1 Ada 1 Akshpreet 1 Alice 1 Adalaine 1 Alabama 38 Alicia 1 Adan 2 Alaina 1 Alicja 1 Adanna 1 Alainah 1 Alicyn 1 Adara 20 Alana 4 Alida 1 Adarah 1 Alanah 2 Aliesha 1 Addisyn 1 Alanda 1 Alifa 1 Adele 1 Alandra 2 Alina 2 Adelle 12 Alanna 1 Aline 1 Adetola 6 Alannah 1 Alinna 1 Adrey 2 Alannis 4 Alisa 1 Adria 1Alara 1 Alisan 9 Adriana 1 Alasha 1 Alisar 6 Adrianna 2 Alaura 23 Alisha 1 Adrianne 1 Alaxandria 2 Alishia 1 Adrien 1 Alayna 1 Alisia 9 Adrienne 1 Alaynna 23 Alison 1 Aerial 1 Alayssia 9 Alissa 1 Aeriel 1 Alberta 1 Alissah 1 Afrika 1 Albertina 1 Alita 4 Aganetha 1 Alea 3 Alix 4 Agatha 2 Aleah 1 Alixandra 2 Agnes 4 Aleasha 4 Aliya 1 Ahmarie 1 Aleashea 1 Aliza 1 Ahnika 7Alecia 1 Allana 2 Aidan 2 Aleena 1 Allannha 1 Aiden 1 Aleeshya 1 Alleah Baby Girl Names Registered in 1996 Page 2 of 28 January, 2006 # Baby Girl Names -

215Th Commencement 05302017
Greetings from the President May 30, 2017 Dear Graduates: Congratulations! You have reached a most significant milestone in your life. Your hard work, determination, and commitment to your education have been rewarded, and you and your loved ones should take pride in your accomplishments and successes. Hunter College certainly takes pride in you. Your Hunter education has prepared you to meet the challenges of a world that is rapidly changing politically, socially, economically, and technologically. As part of the next generation of thoughtful, responsible, and intelligent leaders, you will make a real difference wherever you apply your knowledge and skills. Endless opportunities await you. As you pursue your goals and move forward with your professional and personal lives, please carry with you Hunter's commitment to community, diversity, and service to otheq . We look forward to hearing great things about you, and we hope you will stay connected to the exciting activities and developments on campus. Please remember Hunter College and know that you will always be part of our family. Best wishes for continued success. f4ivtMSincerely, Jennifer J. Raab President 215th Commencement Exercises Presiding: Jennifer J. Raab President Eija Ayravainen Vice President for Student Affairs and Dean ofStudents Opening Ceremony Qin Lin Bachelor ofArts, '17 Processional President's Party and Members of the Faculty Candidates for Degrees National Anthem Carey Renee Anderson Master ofScience in Education, '17 Bagpiper Nicholas M. Rozak Bachelor ofArts, '07 Greetings Sandra Wilkin Board of Trustees of The City University ofNew York Chika Onyejiukwa Student Member, Board of Trustees of The City University ofNew York Charge to the Graduates President Jennifer J. -

1 Noaa /Aslc #18902 1
1 NOAA /ASLC #18902 1 Alaska SeaLife Center Family At the end of the year, the Alaska SeaLife Center w as caring for 202 different species. This brings our grand total to 3,723 individuals. Birds Mammals 13 species, 142 individuals 4 species, 15 individuals --Aviary Birds-- 5 Steller Sea Lions 9 Tufted Puffins 3 Northern Sea Otters 15 Horned Puffins 3 Spotted Seals 2 Black Oystercatchers 4 Ringed Seals 3 Rhinoceros Auklets 4 King Eiders 4 Long-tailed Ducks Fish 2 Harlequin Ducks 62 species, 952 individuals 4 Pigeon Guillemots 2 Smews Invertebrates 14 Red-legged Kittiwakes 123 species, 2,614 individuals 10 Common Murres --Research Birds-- 14 Spectacled Eiders 59 Steller’s Eiders 2 2 From the President and CEO No one was prepared for what 2020 was going to were released at the same time, something never bring. It was the year of the pivot and a year of done at the Center before. extreme uncertainty. In an unprecedented fashion, we closed our doors for two months. Though the If we had a theme for 2020, it would be resiliency. world went into lockdown, the Alaska SeaLife Staff pulled together by cross training, minimizing Center still had animals to feed, a facility to costs, and provided mental support to each other maintain, and animals to rescue and rehabilitate. to get us through uncertain times. I couldn’t be Life behind inside the Center still went on but was prouder of this team. And we were awestruck by certainly different. When we closed our doors in the support of the people of Alaska. -

2021 Summer Season
2021 Summer Season How to use this VIRTUAL program book! Here’s how it works: This program book can be viewed on your computer, tablet, or smartphone, and it has all of the information you need about this summer at BVT! It’s also interactive! That means that there are all sorts of things to click on and learn more about! If you see an advertisement or a logo like this one- Go ahead and give it a click! It’ll take you right to that business’s website or social media page. So you can support those who support BVT! And if you see some blue underlined text, like this right here, that’s a click-able link as well! These links can take you to artists’s websites, local businesses, more BVT content, and all kinds of good stuff. And new things will be added all season long as new shows come to the BVT stage! So scroll through and explore… and enjoy the show! Message from the Artistic Director, Karin Bowersock Places, please. At start time of a theatrical performance, the stage manager gives the places call. “Places, please for the top of the show.” It lets actors and run crew know, it's showtime! It's been a bit of a while since we heard the places call at BVT. I don't need to tell you why. And I can't possibly capture in words how it felt to spend a year without you, with only that tentative—and much less satisfying—virtual connection (Ugh, if I never see another zoom room.....). -

Fiction BOOKS DUE: JANUARY, FEBRUARY, MARCH, APRIL 2013 • Volume 23, Number 1
ROUTE TO: __________________________________ __________________________________ PENGUIN GROUP (USA) __________________________________ Advance Publication Newsletter For Library Managers in Acquisitions and Collection Development Fiction BOOKS DUE: JANUARY, FEBRUARY, MARCH, APRIL 2013 • VOLUME 23, NUMBER 1 A Tale for the TIME BEING RAGE Is BacK THE OFFIce of MercY RUTH OZEKI Adam MANSBACH ARIEL DjaNIKIAN In Tokyo, 16-year-old Nao has decided there’s only “A gutsy act of cultural nostalgia, full of longing for a “If you think a future world without suffering would one escape from her aching loneliness and her class- vanished New York, a chaotic, colorful city full of graf- be a good thing, Ariel Djanikian will convince you to mates’ bullying. But first, she plans to document the fiti and guerilla art. Adam Mansbach is a fearless, reconsider in her impressive debut, The Office of life of her great grandmother, a Buddhist nun. funny, and thoroughly engaging writer.”—Tom Per- Mercy. Gripping, well plotted, and boasting a fasci- A Viking hardcover rotta, author of Little Children. nating setting, this utterly engrossing tale is March • 432 pp. • 978-0-670-02663-0 • $27.95 A Viking hardcover thoughtful and surprising.”—Deborah Harkness, Also available as a Penguin Audiobook January • 304 pp. • 978-0-670-02612-8 • $26.95 author of Shadow of Night and A Discovery of Witches. Digital only, 15.5 hours • Unabridged • 978-1-10-160530-1 • $49.95 Also available as a Penguin Audiobook A Viking hardcover Digital only, 10 hours • Unabridged • 978-1-10-160525-7 -

Baby Girl Names Registered in 2004
Baby Girl Names Registered in 2004 # Baby Girl Names # Baby Girl Names # Baby Girl Names 1 Aafiya 3 Achol 1 Agustina 1 Aaira 2 Ada 1 Ahlam 1 Aalayah 1 Adaiah 1 Ahlia 1 Aaliya 1 Adalee 1 Ahneke 29 Aaliyah 1 Adaleigh 1 Ahstyn 1 Aaliza 1 Adan 1 Aia 1 Aamina 1 Adanna 1 Aicha 1 Aanum 3 Adara 3 Aidan 1 Aarshdeep 1 Adasa 1 Aiden 1 Aasiya 1 Adawncia 1 Aidha 1 Aastha 1 AddahMaria 1 Aiesha 1 Aava 11 Addison 1 Aife 1 Abagail 1 Addisson 1 Aija 1 Abagayl 1 Addyson 1 Aileen 1 Abang 1 Adebola 1 Ailey 1 Abbagael 1 Adelaide 1 Aili 1 Abbagayle 1 Adelaine 2 Ailish 1 Abbegail 1 Adelia 1 Ailsa 12 Abbey 1 Adelina 1 Aime 1 AbbeyLynn 4 Adeline 10 Aimee 4 Abbie 1 Adelle 3 Aimée 7 Abbigail 2 Adelyn 1 Aimeelynn 1 Abbigaile 1 Adelynn 1 Aimi 7 Abbigale 1 Adessah 1 Aine 1 Abbigayle 1 Adia 1 Ainslee 27 Abby 1 Adian 14 Ainsley 5 Abbygail 1 Adina 1 Ainsliegh 1 Abbygale 1 Adison 1 Airael 1 AbbyRuth 2 Aditi 1 Aireana 1 Abegale 1 Adonia 6 Aisha 3 Abigael 1 Adreanna 1 Aishah 125 Abigail 3 Adria 1 AishaSabra 1 Abigale 3 Adrian 1 Aishpreet 8 Abigayle 9 Adriana 2 Aisling 1 Abigial 18 Adrianna 1 Áisling 1 Abinav 2 Adrianne 1 Aislinn 1 Abisha 1 Adrienna 2 Aislyn 1 Abony 3 Adrienne 5 Aislynn 1 Abra 1 Adrina 1 Aiva 1 Abree 2 Adut 1 Aivree 1 Abrial 1 Adyson 1 Aixa 1 Abrianna 1 Aemilie 1 Aiya 1 Abrie 1 Aeries 2 Aiyana 1 Abriel 1 Aerika 2 Aja 2 Abrielle 1 Affia 1 Ajinder 1 Abygale 1 Afrah 1 Ajla 1 Acacia 1 Aftyn 1 Ajock 1 Acadia 1 Agail 2 Akaisha 1 Accalia 3 Aganetha 1 Akakyva 1 Achai 5 Agatha 1 Akara Baby Girl Names Registered in 2004 Page 2 of 36 February, 2004 # Baby Girl Names -

Posthuman Rhetoric and Harris' Sookie Stackhouse Series
California State University, San Bernardino CSUSB ScholarWorks Electronic Theses, Projects, and Dissertations Office of aduateGr Studies 6-2016 COMING OUT OF THE COFFIN AS THE POSTHUMAN: POSTHUMAN RHETORIC AND HARRIS’ SOOKIE STACKHOUSE SERIES Rebecca Ann Garcia California State University - San Bernardino Follow this and additional works at: https://scholarworks.lib.csusb.edu/etd Part of the Other English Language and Literature Commons Recommended Citation Garcia, Rebecca Ann, "COMING OUT OF THE COFFIN AS THE POSTHUMAN: POSTHUMAN RHETORIC AND HARRIS’ SOOKIE STACKHOUSE SERIES" (2016). Electronic Theses, Projects, and Dissertations. 400. https://scholarworks.lib.csusb.edu/etd/400 This Thesis is brought to you for free and open access by the Office of aduateGr Studies at CSUSB ScholarWorks. It has been accepted for inclusion in Electronic Theses, Projects, and Dissertations by an authorized administrator of CSUSB ScholarWorks. For more information, please contact [email protected]. COMING OUT OF THE COFFIN AS THE POSTHUMAN: POSTHUMAN RHETORIC AND HARRIS’ SOOKIE STACKHOUSE SERIES A Thesis Presented to the Faculty of California State University, San Bernardino In Partial Fulfillment of the Requirements for the Degree Master of Arts in English Composition: English Composition/Literature by Rebecca Ann Garcia June 2016 COMING OUT OF THE COFFIN AS THE POSTHUMAN: POSTHUMAN RHETORIC AND HARRIS’ SOOKIE STACKHOUSE SERIES A Thesis Presented to the Faculty of California State University, San Bernardino by Rebecca Garcia June 2016 Approved by: Jacqueline Rhodes, First Reader, English Brenda Glascott, Second Reader, English © 2016 Rebecca Ann Garcia ABSTRACT In this article, I argue that the vampires in Charlaine Harris’ Sookie Stackhouse novels illustrate clearly the posthuman self in its connection beyond itself to other vampires, humans, and non-humans. -

First Name Last Name Honors City St Nation Autumn 2013 Dean's List
AUTUMN 2013 DEAN'S LIST FIRST NAME LAST NAME HONORS CITY ST NATION Jonah Abbey * MISSOULA MT Desiree Acholla * MISSOULA MT Tatem Acree * NAMPA ID Robert Adair III * MISSOULA MT Lorraine Adams ** LIVINGSTON MT Ashley Adams * MISSOULA MT Jessica Adams ** MISSOULA MT Rachel Adams * CHEYENNE WY Molly Adamson * HELENA MT Nathan Adkins * KALISPELL MT Julian Adler * MISSOULA MT Erin Agner ** MISSOULA MT Payton Agnew * SAN DIEGO CA Sadie Ahearn ** MISSOULA MT Tomi Ahearn * MISSOULA MT Lori Ahern * MISSOULA MT Christine Ahlin ** MISSOULA MT Devin Ailport * MISSOULA MT Kuinalu Akane * MISSOULA MT Mackenzie Akins * LAKE ELSINORE CA Amy Albertson * MISSOULA MT Amanda Alexander * MISSOULA MT Abdullah Alhammad * DAMMAM SAUDI ARABIA Emily Allen * CLANCY MT Jacklyn Allen * MISSOULA MT Jason Allen * MISSOULA MT Caroline Allen * TACOMA WA Kelsey Alley * BUTTE MT Samuel Allred * PINESDALE MT Aubrey Allsop ** CORVALLIS MT Connie Allsop * CORVALLIS MT Luke Allsop * MISSOULA MT Marwa Almarhoun * DAMMAM SAUDI ARABIA Erik Alnes * TULSA OK Deborah Alteneder * MISSOULA MT Jacob Altmiller * MISSOULA MT Lillian Alvernaz * GLASGOW MT Jocelyn Ambo * MISSOULA MT Maira Ambris * RAPELJE MT Claire Anchordoqui * DENGUIN FRANCE Mandy Andersen * SAINT IGNATIUS MT Kayli Anderson * BILLINGS MT Amy Anderson * GREAT FALLS MT Andrew Anderson * HELENA MT Courtney Anderson * HELENA MT Kristen Anderson * LIBBY MT Nicole Anderson * MISSOULA MT Samantha Anderson * MISSOULA MT Sara Anderson ** MISSOULA MT Chase Anderson * CHELAN WA Ian Anderson ** DIVERNON IL Kathryn Anderson ** AUCKLAND -

May 2018 Achievers
U.S. and Affiliates, Bermuda and Bahamas 65533 District 1 A RAFAEL ALVARADO PHILIPPE BEAUZILE KISHWAR KHOKHAR ### MOHAMMAD KHOKHAR SHARON MOREY PAM POMPER ### GYLMAR PONCE RAJA RAZZAQ ARACELI RINCON ### ISHRAQ UDDIN ADAM WINSTON JEROME YOUNG ### 65535 District 1 CN ANDREA FORD NATE PASCHAL ### 65536 District 1 CS JERRE PFAFF ### 65537 District 1 D JAMES KLOEPPING RUSSELL SARVER NANCY SHELLHAUSE ### MICHELLE WEIER DENNIS ZIMMERMAN ### 65539 District 1 F RICHARD SHOEMAKER ### 65540 District 1 G RON GILMORE ### 65542 District 1 J SEAN OBRIEN ### 65545 District 2 T1 ERICA GUZMAN SCOTT OWENS #### 65546 District 2 T2 TOMMY OGDEN #### Thursday, June 27, 2019 Page 1 of 77 Diamond Centennial Awards U.S. and Affiliates, Bermuda and Bahamas 65547 District 2 T3 ALAN NICHOLAS VICTOR RAMIREZ JUDI VERSLYPE #### 65548 District 2 E1 TOM BOYD RONALD DUNCAN TERESA ECHOLS #### 65549 District 2 E2 CHARLES BETHARDS JOHN PAUL BURNETT MINDY MONROE #### MARK WOOD #### 65550 District 2 X1 MARISSA BUTTS MATT KEEVER JOSEPH KYLE #### BOB LEE CHARLES THOMPSON ALICIA VELASCO #### 65551 District 2 X2 EARNEST LARGENT CASSANDRA SAMPSON C LARRY SMITH #### HAROLD WIESE #### 65552 District 2 X3 JACK BRADLEY RUSS D COCHRAN EDWARD HUESKE #### BOBBY VIRGEN-SIMS #### 65553 District 2 A1 SYLVIA CLANTON A J DOLLE DAVE RICHISON #### SCOTT SPOONTS #### 65554 District 2 A2 ALBERT GARCIA FELIPE GARZA ANTONIO GONZALES #### DONALD KIRCHHOFF RODNEY MARTINEZ ESMERALDA RODRIGUEZ #### 65555 District 2 A3 ANTONIO GARCIA SAM GONZALEZ MARY RUIZ #### VIRGINIA SMITH RICHARD TALBERT #### 65556 -

Annual Report 2005-2006
Annual Report 2005-2006 PrincetonUniversity Office of Information Technology Annual Report 2005-2006 PrincetonUniversity 2005-2006 Annual Report Page 1 Produced by the staff of the Office of Information Technology. Office of Information Technology, 315 Nassau Hall, Princeton University, Princeton, NJ 08544. Copyright © 2006, Office of Information Technology, Princeton University. All rights reserved. Printed by the Office of Printing and Mailing, Princeton University. Page 2 Office of Information Technology Table of Contents Letter from the Vice President ..................................................................................... 5 OIT Mission and Goals ............................................................................................... 7 FY06 OIT Timeline ...................................................................................................... 8 OIT by the Numbers, FY06 ....................................................................................... 12 OIT Organization ...................................................................................................... 13 IT Decision-Making Process Map ............................................................................ 14 Senior Advisory Group on IT.................................................................................... 15 Administrative Systems Planning Group .................................................................. 16 Project Managers Team ............................................................................................ -

Seniors & Disabled
HELPING PEOPLE HELP THEMSELVES WINTER 2012 Joyce Seely touches the lives of seniors & disabled n December 5, Joyce other items. always enjoyed the company of Seely delivered 152 This152 year, her donation was in seniors and too often finds that Ogift-filled shoeboxes memory of her mother who they have become the forgotten. to ACCESS to be delivered recently passed away. “I feel they should be honored for to our seniors and disabled all they have gone through clients during the holidays. and survived. This is my “This is my way of letting way of letting them know She is busy all through the year them know someone is someone is thinking of assembling those boxes. A few them.” of her friends and her sister help thinking of them.” Joyce’s original volunteer purchase items, put the boxes history was with animal together and wrap them. She “I actually started doing this organizations. She has always includes items that she a couple of years ago as my volunteered at Jackson County makes herself, such as lavender mother’s health was failing and Animal Care and Control and she sachets, coffee mugs, gloves, I was taking care of her,” she and a friend even started Spay puzzle books, pens and pencils, says. “I often felt helpless to stop and Neuter Your Pet and Animals small stuffed animals, photo what was happening to her, but in Distress. albums and frames, candy, and she loved the idea of the senior “The last few years I shifted my shoeboxes focus to ACCESS after discovering in this issue: and we how important they were to • Brighten someone’s life with a shoebox • Special program to help feed seniors shared those caring for someone who is • Message from our Executive Director. -
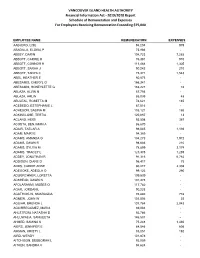
2019/2020 Report Schedule of Remuneration and Expenses for Employees Receiving Remuneration Exceeding $75,000
VANCOUVER ISLAND HEALTH AUTHORITY Financial Information Act - 2019/2020 Report Schedule of Remuneration and Expenses For Employees Receiving Remuneration Exceeding $75,000 EMPLOYEE NAME REMUNERATION EXPENSES AABJERG, LISE 94,204 979 ABADILLA, ELOISA P 75,966 - ABBEY, DARIN 104,723 7,265 ABBOTT, CARRIE K 76,381 910 ABBOTT, CONNOR H 111,468 1,305 ABBOTT, SARAH J 90,242 210 ABBOTT, TANYA C 75,971 1,543 ABEL, HEATHER E 80,873 - ABESAMIS, CHERYL G 166,341 - ABESAMIS, HONEYLETTE G 153,227 78 ABLAZA, ALVIN N 87,756 - ABLAZA, ARLIN 85,099 48 ABULEAL, ROSETTA M 78,621 185 ACEBEDO, ESTEPHANIE L 87,516 - ACHESON, SABINA M 103,121 186 ACKINCLOSE, TERTIA 120,887 13 ACLAND, HEIDI 93,598 367 ACOSTA, BENJAMIN A 86,670 - ADAIR, TAELAR A 98,045 1,193 ADAM, MAIRI E 94,389 - ADAMS, AMANDA G 104,273 1,972 ADAMS, DAWN R 98,686 210 ADAMS, SYLVIA M 78,899 2,174 ADAMS, TRACEY L 123,375 1,259 ADDEY, JONATHAN R 91,316 6,782 ADDISON, DIANE D 96,417 75 ADDS, CARRIE ANNE 80,217 2,308 ADEGOKE, ADEOLA O 99,123 290 ADLKIRCHNER, LORETTA 109,539 - ADSHEAD, DAWN N 101,378 - AFOLARANMI, MOSES O 117,782 - AGAR, JORDAN L 90,223 - AGATHOKLIS, SMARAGDA 76,880 778 AGNEW, JOHN W 101,506 22 AGUIAR, BRENDA L 101,784 2,043 AGUIRREGOMEZ, MARIA 89,004 - AHLSTROM, NATASHA D 92,786 - AHLUWALIA, SANGEETA 146,507 - AHMED, SAMINA K 75,248 1,460 AIERS, JENNIFER E 84,546 606 AIKMAN, KRISTY L 85,051 192 AIRD, WENDY 101,878 - AITCHISON, DEBBORAH L 92,059 - AITKEN, SANDRA V 94,624 - VANCOUVER ISLAND HEALTH AUTHORITY Financial Information Act - 2019/2020 Report Schedule Of Remuneration And Expenses