Intelligent Microarray Data Analysis Through Non-Negative Matrix Factorization to Study Human Multiple Myeloma Cell Lines
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ACOX3 Antibody
Product Datasheet ACOX3 antibody Catalog No: #22136 Orders: [email protected] Description Support: [email protected] Product Name ACOX3 antibody Host Species Rabbit Clonality Polyclonal Purification Purified by antigen-affinity chromatography. Applications WB IHC Species Reactivity Hu Immunogen Type Recombinant protein Immunogen Description Recombinant protein fragment contain a sequence corresponding to a region within amino acids 408 and 613 of ACOX3 Target Name ACOX3 Accession No. Swiss-Prot:O15254Gene ID:8310 Concentration 1mg/ml Formulation Supplied in 0.1M Tris-buffered saline with 20% Glycerol (pH7.0). 0.01% Thimerosal was added as a preservative. Storage Store at -20°C for long term preservation (recommended). Store at 4°C for short term use. Application Details Predicted MW: 70kd Western blotting: 1:500-1:3000 Immunohistochemistry: 1:100-1:500 Images Sample (30 ug of whole cell lysate) A: A549 7.5% SDS PAGE Primary antibody diluted at 1: 1000 Address: 8400 Baltimore Ave., Suite 302, College Park, MD 20740, USA http://www.sabbiotech.com 1 Immunohistochemical analysis of paraffin-embedded H441 xenograft, using ACOX3 antibody at 1: 500 dilution. Background Acyl-Coenzyme A oxidase 3 also know as pristanoyl -CoA oxidase (ACOX3)is involved in the desaturation of 2-methyl branched fatty acids in peroxisomes. Unlike the rat homolog, the human gene is expressed in very low amounts in liver such that its mRNA was undetectable by routine Northern-blot analysis or its product by immunoblotting or by enzyme activity measurements. However the human cDNA encoding a 700 amino acid protein with a peroxisomal targeting C-terminal tripeptide S-K-L was isolated and is thought to be expressed under special conditions such as specific developmental stages or in a tissue specific manner in tissues that have not yet been examined. -

ACOX3 Human Shrna Plasmid Kit (Locus ID 8310) Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TL314988 ACOX3 Human shRNA Plasmid Kit (Locus ID 8310) Product data: Product Type: shRNA Plasmids Product Name: ACOX3 Human shRNA Plasmid Kit (Locus ID 8310) Locus ID: 8310 Vector: pGFP-C-shLenti (TR30023) Format: Lentiviral plasmids Components: ACOX3 - Human, 4 unique 29mer shRNA constructs in lentiviral GFP vector(Gene ID = 8310). 5µg purified plasmid DNA per construct Non-effective 29-mer scrambled shRNA cassette in pGFP-C-shLenti Vector, TR30021, included for free. RefSeq: NM_001101667, NM_003501, NM_003501.1, NM_003501.2, NM_001101667.1, BC017053, NM_003501.3, NM_001101667.2 Summary: Acyl-Coenzyme A oxidase 3 also know as pristanoyl -CoA oxidase (ACOX3)is involved in the desaturation of 2-methyl branched fatty acids in peroxisomes. Unlike the rat homolog, the human gene is expressed in very low amounts in liver such that its mRNA was undetectable by routine Northern-blot analysis or its product by immunoblotting or by enzyme activity measurements. However the human cDNA encoding a 700 amino acid protein with a peroxisomal targeting C-terminal tripeptide S-K-L was isolated and is thought to be expressed under special conditions such as specific developmental stages or in a tissue specific manner in tissues that have not yet been examined. [provided by RefSeq, Jul 2008] shRNA Design: These shRNA constructs were designed against multiple splice variants at this gene locus. To be certain that your variant of interest is targeted, please contact [email protected]. -

ACOX3 (H-1): Sc-390624
SAN TA C RUZ BI OTEC HNOL OG Y, INC . ACOX3 (H-1): sc-390624 BACKGROUND APPLICATIONS ACOX3 (acyl-Coenzyme A oxidase 3), also known as BRCOX or PRCOX, is a ACOX3 (H-1) is recommended for detection of ACOX3 of mouse, rat and 700 amino acid protein that localizes to peroxisomes and belongs to the acyl- human origin by Western Blotting (starting dilution 1:100, dilution range CoA oxidase family. Using FAD as a cofactor, ACOX3 catalyzes the desatura - 1:100-1:1000), immunoprecipitation [1-2 µg per 100-500 µg of total protein tion of 2-methyl branched fatty acids in peroxisomes, thereby playing an impor - (1 ml of cell lysate)], immunofluorescence (starting dilution 1:50, dilution tant role in peroxisomal fatty acid β-oxidation. Human ACOX3 shares 75% range 1:50-1:500) and solid phase ELISA (starting dilution 1:30, dilution sequence identity with its rat counterpart, suggesting a conserved role range 1:30-1:3000). between species. Multiple isoforms of ACOX3 exist due to alternative splic - Suitable for use as control antibody for ACOX3 siRNA (h): sc-89236, ACOX3 ing events. The gene encoding ACOX3 maps to human chromosome 4, which siRNA (m): sc-140819, ACOX3 shRNA Plasmid (h): sc-89236-SH, ACOX3 encodes nearly 6% of the human genome and has the largest gene deserts shRNA Plasmid (m): sc-140819-SH, ACOX3 shRNA (h) Lentiviral Particles: (regions of the genome with no protein encoding genes) of all of the human sc-89236-V and ACOX3 shRNA (m) Lentiviral Particles: sc-140819-V. chromosomes. Defects in some of the genes located on chromosome 4 are associated with Huntington’s disease, Ellis-van Creveld syndrome, methyl - Molecular Weight of ACOX3: 78 kDa. -

Novel and Highly Recurrent Chromosomal Alterations in Se´Zary Syndrome
Research Article Novel and Highly Recurrent Chromosomal Alterations in Se´zary Syndrome Maarten H. Vermeer,1 Remco van Doorn,1 Remco Dijkman,1 Xin Mao,3 Sean Whittaker,3 Pieter C. van Voorst Vader,4 Marie-Jeanne P. Gerritsen,5 Marie-Louise Geerts,6 Sylke Gellrich,7 Ola So¨derberg,8 Karl-Johan Leuchowius,8 Ulf Landegren,8 Jacoba J. Out-Luiting,1 Jeroen Knijnenburg,2 Marije IJszenga,2 Karoly Szuhai,2 Rein Willemze,1 and Cornelis P. Tensen1 Departments of 1Dermatology and 2Molecular Cell Biology, Leiden University Medical Center, Leiden, the Netherlands; 3Department of Dermatology, St Thomas’ Hospital, King’s College, London, United Kingdom; 4Department of Dermatology, University Medical Center Groningen, Groningen, the Netherlands; 5Department of Dermatology, Radboud University Nijmegen Medical Center, Nijmegen, the Netherlands; 6Department of Dermatology, Gent University Hospital, Gent, Belgium; 7Department of Dermatology, Charite, Berlin, Germany; and 8Department of Genetics and Pathology, Rudbeck Laboratory, University of Uppsala, Uppsala, Sweden Abstract Introduction This study was designed to identify highly recurrent genetic Se´zary syndrome (Sz) is an aggressive type of cutaneous T-cell alterations typical of Se´zary syndrome (Sz), an aggressive lymphoma/leukemia of skin-homing, CD4+ memory T cells and is cutaneous T-cell lymphoma/leukemia, possibly revealing characterized by erythroderma, generalized lymphadenopathy, and pathogenetic mechanisms and novel therapeutic targets. the presence of neoplastic T cells (Se´zary cells) in the skin, lymph High-resolution array-based comparative genomic hybridiza- nodes, and peripheral blood (1). Sz has a poor prognosis, with a tion was done on malignant T cells from 20 patients. disease-specific 5-year survival of f24% (1). -

Regions of Homozygosity Identified by SNP Microarray Analysis Aid in The
ORIGINAL RESEARCH ARTICLE ©American College of Medical Genetics and Genomics Regions of homozygosity identified bySN P microarray analysis aid in the diagnosis of autosomal recessive disease and incidentally detect parental blood relationships Kristen Lipscomb Sund, PhD, MS1, Sarah L. Zimmerman, PhD1, Cameron Thomas, MD2, Anna L. Mitchell, MD, PhD3, Carlos E. Prada, MD1, Lauren Grote, BS1, Liming Bao, MD, PhD1, Lisa J. Martin, PhD1 and Teresa A. Smolarek, PhD1 Purpose: The purpose of this study was to document the ability of was suspected in the parents of at least 11 patients with regions of single-nucleotide polymorphism microarray to identify copy-neutral homozygosity covering >21.3% of their autosome. In four patients regions of homozygosity, demonstrate clinical utility of regions of from two families, homozygosity mapping discovered a candidate homozygosity, and discuss ethical/legal implications when regions of gene that was sequenced to identify a clinically significant mutation. homozygosity are associated with a parental blood relationship. Conclusion: This study demonstrates clinical utility in the identifica- Methods: Study data were compiled from consecutive samples tion of regions of homozygosity, as these regions may aid in diagnosis sent to our clinical laboratory over a 3-year period. A cytogenetics of the patient. This study establishes the need for careful reporting, database identified patients with at least two regions of homozygosity thorough pretest counseling, and careful electronic documentation, >10 Mb on two separate chromosomes. A chart review was conduct- as microarray has the capability of detecting previously unknown/ ed on patients who met the criteria. unreported relationships. Results: Of 3,217 single-nucleotide polymorphism microarrays, Genet Med 2013:15(1):70–78 59 (1.8%) patients met inclusion criteria. -

Downloaded from Ftp://Ftp.Uniprot.Org/ on July 3, 2019) Using Maxquant (V1.6.10.43) Search Algorithm
bioRxiv preprint doi: https://doi.org/10.1101/2020.11.17.385096; this version posted November 17, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. The proteomic landscape of resting and activated CD4+ T cells reveal insights into cell differentiation and function Yashwanth Subbannayya1, Markus Haug1, Sneha M. Pinto1, Varshasnata Mohanty2, Hany Zakaria Meås1, Trude Helen Flo1, T.S. Keshava Prasad2 and Richard K. Kandasamy1,* 1Centre of Molecular Inflammation Research (CEMIR), and Department of Clinical and Molecular Medicine (IKOM), Norwegian University of Science and Technology, N-7491 Trondheim, Norway 2Center for Systems Biology and Molecular Medicine, Yenepoya (Deemed to be University), Mangalore, India *Correspondence to: Professor Richard Kumaran Kandasamy Norwegian University of Science and Technology (NTNU) Centre of Molecular Inflammation Research (CEMIR) PO Box 8905 MTFS Trondheim 7491 Norway E-mail: [email protected] (Kandasamy R K) Tel.: +47-7282-4511 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.11.17.385096; this version posted November 17, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract CD4+ T cells (T helper cells) are cytokine-producing adaptive immune cells that activate or regulate the responses of various immune cells. -

Peroxisomal Lactate Dehydrogenase Is Generated by Translational Readthrough in Mammals
1 Peroxisomal lactate dehydrogenase is generated 2 by translational readthrough in mammals 3 4 Fabian Schueren1†, Thomas Lingner2†, Rosemol George1†, Julia Hofhuis1, 5 Corinna Dickel1, Jutta Gärtner1*, Sven Thoms1* 6 7 1Department of Pediatrics and Adolescent Medicine, University Medical Center, 8 Georg-August-University Göttingen, Germany 9 2Department of Bioinformatics, Institute for Microbiology and Genetics, 10 Georg-August-University Göttingen, Germany 11 † Co-first author 12 13 * Correpondence: [email protected] (J.G.); [email protected] 14 (S.T.) 15 16 Competing interest statement: The authors declare no competing interest. 17 18 ABSTRACT 19 Translational readthrough gives rise to low abundance proteins with C-terminal 20 extensions beyond the stop codon. To identify functional translational readthrough, we 21 estimated the readthrough propensity (RTP) of all stop codon contexts of the human 22 genome by a new regression model in silico, identified a nucleotide consensus motif for 23 high RTP by using this model, and analyzed all readthrough extensions in silico with a 24 new predictor for peroxisomal targeting signal type 1 (PTS1). Lactate dehydrogenase B 25 (LDHB) showed the highest combined RTP and PTS1 probability. Experimentally we 26 show that at least 1.6% of the total cellular LDHB getting targeted to the peroxisome by 27 a conserved hidden PTS1. The readthrough-extended lactate dehydrogenase subunit 28 LDHBx can also co-import LDHA, the other LDH subunit into peroxisomes. Peroxisomal 29 LDH is conserved in mammals and likely contributes to redox equivalent regeneration in 30 peroxisomes. 31 1 32 33 INTRODUCTION 34 Translation of genetic information encoded in mRNAs into proteins is carried out by ribosomes. -

Content Based Search in Gene Expression Databases and a Meta-Analysis of Host Responses to Infection
Content Based Search in Gene Expression Databases and a Meta-analysis of Host Responses to Infection A Thesis Submitted to the Faculty of Drexel University by Francis X. Bell in partial fulfillment of the requirements for the degree of Doctor of Philosophy November 2015 c Copyright 2015 Francis X. Bell. All Rights Reserved. ii Acknowledgments I would like to acknowledge and thank my advisor, Dr. Ahmet Sacan. Without his advice, support, and patience I would not have been able to accomplish all that I have. I would also like to thank my committee members and the Biomed Faculty that have guided me. I would like to give a special thanks for the members of the bioinformatics lab, in particular the members of the Sacan lab: Rehman Qureshi, Daisy Heng Yang, April Chunyu Zhao, and Yiqian Zhou. Thank you for creating a pleasant and friendly environment in the lab. I give the members of my family my sincerest gratitude for all that they have done for me. I cannot begin to repay my parents for their sacrifices. I am eternally grateful for everything they have done. The support of my sisters and their encouragement gave me the strength to persevere to the end. iii Table of Contents LIST OF TABLES.......................................................................... vii LIST OF FIGURES ........................................................................ xiv ABSTRACT ................................................................................ xvii 1. A BRIEF INTRODUCTION TO GENE EXPRESSION............................. 1 1.1 Central Dogma of Molecular Biology........................................... 1 1.1.1 Basic Transfers .......................................................... 1 1.1.2 Uncommon Transfers ................................................... 3 1.2 Gene Expression ................................................................. 4 1.2.1 Estimating Gene Expression ............................................ 4 1.2.2 DNA Microarrays ...................................................... -
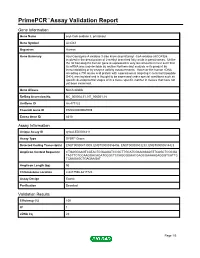
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name acyl-CoA oxidase 3, pristanoyl Gene Symbol ACOX3 Organism Human Gene Summary Acyl-Coenzyme A oxidase 3 also know as pristanoyl -CoA oxidase (ACOX3)is involved in the desaturation of 2-methyl branched fatty acids in peroxisomes. Unlike the rat homolog the human gene is expressed in very low amounts in liver such that its mRNA was undetectable by routine Northern-blot analysis or its product by immunoblotting or by enzyme activity measurements. However the human cDNA encoding a 700 amino acid protein with a peroxisomal targeting C-terminal tripeptide S-K-L was isolated and is thought to be expressed under special conditions such as specific developmental stages or in a tissue specific manner in tissues that have not yet been examined. Gene Aliases Not Available RefSeq Accession No. NC_000004.11, NT_006051.18 UniGene ID Hs.479122 Ensembl Gene ID ENSG00000087008 Entrez Gene ID 8310 Assay Information Unique Assay ID qHsaCED0004211 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000413009, ENST00000356406, ENST00000503233, ENST00000514423 Amplicon Context Sequence CTGAGGAAGTCATACTCGAAGATCCGCTTGCATCGAAGGAAGTTCAGCTCGCGA TACTTCTCCAAGGACAGATCGGCTCCAGGGGAACGAGCGAAAAGAGGGTCATTC TCAAGAGCTGAGAAGAT Amplicon Length (bp) 95 Chromosome Location 4:8417596-8417720 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 100 R2 1 cDNA Cq 22 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 83.5 gDNA Cq 23.36 Specificity (%) 100 Information to assist with data -
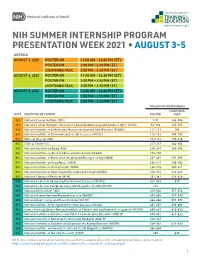
Summer Presentation Week
NIH SUMMER INTERNSHIP PROGRAM PRESENTATION WEEK 2021 • AUGUST 3-5 AGENDA AUGUST 3, 2021 POSTER AM 11:00 AM – 12:30 PM (ET) POSTER PM 1:00 PM – 2:30 PM (ET) LIGHTNING TALK 3:00 PM – 4:30 PM (ET) AUGUST 4, 2021 POSTER AM 11:00 AM – 12:30 PM (ET) POSTER PM 1:00 PM – 2:30 PM (ET) LIGHTNING TALK 3:00 PM – 4:30 PM (ET) AUGUST 5, 2021 POSTER AM 11:00 AM – 12:30 PM (ET) POSTER PM 1:00 PM – 2:30 PM (ET) LIGHTNING TALK 3:00 PM – 4:30 PM (ET) PRESENTATION NUMBERS LIGHTNING DATE INSTITUTE OR CENTER POSTER TALK 8/3 National Cancer Institute (NCI) 1-91 144-184 8/3 National Cancer Institute – Division of Cancer Epidemiology and Genetics (NCI – DCEG) 92-110 185-187 8/3 National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) 111-113 188 8/3 National Institute of Environmental Health Sciences (NIEHS) 114-141 189-192 8/3 Office of Director (OD) 142-143 193-216 8/4 Clinical Center (CC) 217-237 362-364 8/4 National Institute on Aging (NIA) 238-247 365-396 8/4 National Institute on Alcohol Abuse and Alcoholism (NIAAA) 248-256 8/4 National Institute of Biomedical Imaging and Bioengineering (NIBIB) 257-261 397-399 8/4 National Institute on Drug Abuse (NIDA) 262-279 400-402 8/4 National Institute of Mental Health (NIMH) 280-315 403-411 8/4 National Institute of Neurological Disorders and Stroke (NINDS) 316-351 412-423 8/4 National Library of Medicine (NLM) 352-361 424-426 8/5 National Center for Advancing Translational Sciences (NCATS) 427-433 570 8/5 National Center for Complementary and Integrative Health (NCCIH) 434 8/5 National Eye Institute -

Protein Lysine Crotonylation: Past, Present, Perspective ✉ ✉ Gaoyue Jiang1,3, Chunxia Li2,3, Meng Lu2,3, Kefeng Lu 2 and Huihui Li 1
www.nature.com/cddis REVIEW ARTICLE OPEN Protein lysine crotonylation: past, present, perspective ✉ ✉ Gaoyue Jiang1,3, Chunxia Li2,3, Meng Lu2,3, Kefeng Lu 2 and Huihui Li 1 © The Author(s) 2021 Lysine crotonylation has been discovered in histone and non-histone proteins and found to be involved in diverse diseases and biological processes, such as neuropsychiatric disease, carcinogenesis, spermatogenesis, tissue injury, and inflammation. The unique carbon–carbon π-bond structure indicates that lysine crotonylation may use distinct regulatory mechanisms from the widely studied other types of lysine acylation. In this review, we discussed the regulation of lysine crotonylation by enzymatic and non-enzymatic mechanisms, the recognition of substrate proteins, the physiological functions of lysine crotonylation and its cross- talk with other types of modification. The tools and methods for prediction and detection of lysine crotonylation were also described. Cell Death and Disease (2021) 12:703 ; https://doi.org/10.1038/s41419-021-03987-z INTRODUCTION transcriptional activation [10]. Structurally, Kcr is four-carbon in Protein posttranslational modifications (PTMs) are important length and the crotonyl modification contains a carbon–carbon epigenetic regulatory mechanisms involved in diverse biological (C–C) π-bond that results in a unique rigid planar conformation [1]. processes, such as DNA replication, transcription, cell differentia- In this review, we will discuss the enzymatic and non-enzymatic tion, and organismal development. Dysregulation of PTMs is regulation of crotonylation, the cellular and physiological func- associated with a number of diseases, e.g., neuropsychiatric tions of Kcr, the cross-talk between Kcr with other PTMs, and the disease, carcinogenesis, and tissue injury [1]. -

Quantitative Proteomic Analysis of Low Linolenic Acid Transgenic Soybean Reveals Perturbations of Fatty Acid Metabolic Pathways
RESEARCH ARTICLE Mass Spectrometry www.proteomics-journal.com Quantitative Proteomic Analysis of Low Linolenic Acid Transgenic Soybean Reveals Perturbations of Fatty Acid Metabolic Pathways Nazrul Islam, Philip D. Bates, K. M. Maria John, Hari B. Krishnan, Zhanyuan J. Zhang, Devanand L. Luthria, and Savithiry S. Natarajan* modifications, gene manipulations To understand the effect of fatty acid desaturase gene (GmFAD3) silencing on for oil composition were intensively perturbation of fatty acid (FA) metabolic pathways, the changes are compared investigated[1,2] due to consumer health in protein profiling in control and low linolenic acid transgenic soybeans using awareness and increased demand for tandem mass tag based mass spectrometry. Protein profiling of the transgenic vegetable oil. Soybean oil constitutes about 60% of total world seed oil (http:// line unveiled changes in several key enzymes of FA metabolism. This includes www.soystats.com) and most of it is used enzymes of lower abundance; fabH, fabF, and thioestrase associated with FA for human consumption. initiation, elongation, and desaturation processes and LOX1 5, ACOX, The fatty acid composition of the ACAA1, MFP2 associated with β-oxidation of α-linolenic acids pathways. In soybean oil is approximately 13% of addition, the GmFAD3 silencing results in a significant reduction in one of the palmitic acid (16:0), 4% stearic acid (18:0), 20% oleic acid (18:1), 55% linoleic major allergens, Gly m 4 (C6T3L5). These results are important for exploring acid (18:2), and 7–10% linolenic acid how plants adjust in their biological processes when certain changes are (18:3).[3] The palmitic and stearic acids induced in the genetic makeup.