Multimetallic Alkaline Earth Hydride Cations OM Revsion.PDF
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Densifying Metal Hydrides with High Temperature and Pressure
3,784,682 United States Patent Office Patented Jan. 8, 1974 feet the true density. That is, by this method only theo- 3,784,682 retical or near theoretical densities can be obtained by DENSIFYING METAL HYDRIDES WITH HIGH making the material quite free from porosity (p. 354). TEMPERATURE AND PRESSURE The true density remains the same. Leonard M. NiebylsM, Birmingham, Mich., assignor to Ethyl Corporation, Richmond, Va. SUMMARY OF THE INVENTION No Drawing. Continuation-in-part of abandoned applica- tion Ser. No. 392,370, Aug. 24, 1964. This application The process of this invention provides a practical Apr. 9,1968, Ser. No. 721,135 method of increasing the true density of hydrides of Int. CI. COlb 6/00, 6/06 metals of Groups II-A, II-B, III-A and III-B of the U.S. CI. 423—645 8 Claims Periodic Table. More specifically, true densities of said 10 metal hydrides may be substantially increased by subject- ing a hydride to superatmospheric pressures at or above ABSTRACT OF THE DISCLOSURE fusion temperatures. When beryllium hydride is subjected A method of increasing the density of a hydride of a to this process, a material having a density of at least metal of Groups II-A, II-B, III-A and III-B of the 0.69 g./cc. is obtained. It may or may not be crystalline. Periodic Table which comprises subjecting a hydride to 15 a pressure of from about 50,000 p.s.i. to about 900,000 DESCRIPTION OF THE PREFERRED p.s.i. at or above the fusion temperature of the hydride; EMBODIMENT i.e., between about 65° C. -

(12) United States Patent (10) Patent No.: US 9.440,228 B2 H0s0no Et Al
USOO9440228B2 (12) United States Patent (10) Patent No.: US 9.440,228 B2 H0s0no et al. (45) Date of Patent: Sep. 13, 2016 (54) PEROVSKITE OXIDE CONTAINING (58) Field of Classification Search HYDRIDE ION, AND METHOD FOR CPC ........................................................ BO1J 31 f(00 MANUFACTURING SAME See application file for complete search history. (75) Inventors: Hideo Hosono, Tokyo (JP); Hiroshi (56) References Cited Kageyama, Kyoto (JP); Yoji Kobayashi, Kyoto (JP); Mikio Takano, Kyoto (JP): Takeshi Yajima, Kyoto FOREIGN PATENT DOCUMENTS (JP) JP 2005-100978 A 4/2005 JP 2006-199578 A 8, 2006 (73) Assignee: JAPAN SCIENCE AND TECHNOLOGY AGENCY, (Continued) Kawaguchi-shi (JP) OTHER PUBLICATIONS (*) Notice: Subject to any disclaimer, the term of this Kobayashi et al(An oxyhydride of BaTiO3 exhibiting hydride patent is extended or adjusted under 35 exchange and electronic conductivity, Nature Materials, vol. 11, U.S.C. 154(b) by 245 days. (2012) pp. 507-511 published online on Apr. 15, 2012).* (21) Appl. No.: 14/130,184 (Continued) (22) PCT Filed: Jul. 5, 2012 Primary Examiner — Melvin C Mayes (86). PCT No.: PCT/UP2012/067.157 Assistant Examiner — Michael Forrest (74) Attorney, Agent, or Firm — Westerman, Hattori, S 371 (c)(1), Daniels & Adrian, LLP (2), (4) Date: Dec. 30, 2013 (87) PCT Pub. No.: WO2013/008705 (57) ABSTRACT Problem Many oxide-ion conductors exhibit high func PCT Pub. Date: Jan. 17, 2013 tionality at high temperatures due to the large weight and (65) Prior Publication Data charge of oxide ions, and it has been difficult to achieve the functionality at low temperatures. US 2014/O128252 A1 May 8, 2014 Solution. -

Bacitracin B:0050 Medical Surveillance: Evaluation by a Qualified Allergist
B Bacitracin B:0050 Medical Surveillance: Evaluation by a qualified allergist. Kidney function tests. First Aid: In case of large-scale exposure, the directions for Molecular Formula: C H N O S 66 103 17 16 medicines (nonspecific, n.o.s.) would be applied as follows: Synonyms: Ayfivin; Baciguent; Baci-Jel; Baciliquin; Move victim to fresh air; call emergency medical care. If Bacitek ointment; Fortracin; Parentracin; Penitracin; not breathing, give artificial respiration. If breathing is diffi- Topitracin; Zutracin cult, give oxygen. In case of contact with material, immedi- CAS Registry Number: 1405-87-4 ® ately flush skin or eyes with running water for at least RTECS Number: CP0175000 15 min. Speed in removing material from skin is of extreme UN/NA & ERG Number: UN3249 (medicine, solid, toxic, importance. Remove and isolate contaminated clothing and n.o.s.)/151 shoes at the site. Keep victim quiet and maintain normal EC Number: 215-786-2 body temperature. Effects may be delayed; keep victim Regulatory Authority and Advisory Bodies under observation. Listed on the TSCA inventory. Storage: Color Code—Green: General storage may be used. List of Acutely Toxic Chemicals, Chemical Emergency Shipping: The DOT category of medicine, solid, toxic, n.o.s. Preparedness Program (EPA) and formerly on CERCLA/ calls for the label of “POISONOUS/TOXIC MATERIALS.” SARA 40CFR302, Table 302.4 Extremely Hazardous Bacitracin would fall in Hazard Class 6.1 and in Packing Substances List. Dropped from listing in 1988. Group III. Listed on Canada’s DSL List. Spill Handling: Evacuate and restrict persons not wearing WGK (German Aquatic Hazard Class): No value assigned. -

Structure and Hydrogen Dynamics of Alkaline Earth Metal Hydrides Investigated with Neutron Scattering
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 5-2020 Structure and Hydrogen Dynamics of Alkaline Earth Metal Hydrides Investigated with Neutron Scattering Eric Christopher Novak University of Tennessee, [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Recommended Citation Novak, Eric Christopher, "Structure and Hydrogen Dynamics of Alkaline Earth Metal Hydrides Investigated with Neutron Scattering. " PhD diss., University of Tennessee, 2020. https://trace.tennessee.edu/utk_graddiss/5842 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Eric Christopher Novak entitled "Structure and Hydrogen Dynamics of Alkaline Earth Metal Hydrides Investigated with Neutron Scattering." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the equirr ements for the degree of Doctor of Philosophy, with a major in Materials Science and Engineering. Takeshi Egami, Major Professor We have read this dissertation and recommend its acceptance: Niina Jalarvo, David Keffer, Katharine Page Accepted for the Council: Dixie L. Thompson Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) Structure and Hydrogen Dynamics of Alkaline Earth Metal Hydrides Investigated with Neutron Scattering A Dissertation Presented for the Doctor of Philosophy Degree The University of Tennessee, Knoxville Eric Christopher Novak May 2020 Copyright © 2020 by Eric Christopher Novak All rights reserved. -

Ab Initio Adiabatic Study of the Agh System Tahani A
www.nature.com/scientificreports OPEN Ab initio adiabatic study of the AgH system Tahani A. Alrebdi1, Hanen Souissi2*, Fatemah H. Alkallas1 & Fatma Aouaini1 In the framework of the Born–Oppenheimer (BO) method, we illustrate our ab-initio spectroscopic study of the of silver hydride molecule. The calculation of 48 electrons for this system is very difcult, so we have been employed a pseudo-potential (P.P) to reduce the big number of electrons to two electrons of valence, which is proposed by Barthelat and Durant. This allowed us to make a confguration interaction (CI). The potential energy curves (PECs) and the spectroscopic constants of AgH have been investigated for Σ+, Π and Δ symmetries. We have been determined the permanent and transition dipole moments (PDM and TDM), the vibrational energies levels and their spacing. We compared our results with the available experimental and theoretical results in the literature. We found a good accordance with the experimental and theoretical data that builds a validation of the choice of our approach. Transition metal hydrides (TMH) play a crucial role in the chemistry due to their potential use from catalysis to energy applications1–3. In this context, many eforts have been carried out understand their spectroscopic, electronic, and structural properties of TMH. Among them, AgH have been studied by Stephen et al.4 using large valence basis sets in connective with relativistic efective core potentials (RECPs). Te silver hydride AgH molecule has been the subject of several experimental5–11 and theoretical4, 12–25 works. Tere is four experimental studies have been examined by Le Roy et al.5, Seto et al.6, Rolf-Dieter et al.7 and Helmut et al.8. -

Chemistry of Alkaline Earth Metals: It Is Not All Ionic and Definitely Not Boring!
3XEOLVKHGLQ&RRUGLQDWLRQ&KHPLVWU\5HYLHZV ZKLFKVKRXOGEHFLWHGWRUHIHUWRWKLVZRUN Chemistry of alkaline earth metals: It is not all ionic and definitely not boring! Katharina M. Fromm University of Fribourg, Department of Chemistry, Chemin du Musée 9, CH-1700 Fribourg, Switzerland Some scientists might consider group 2 chemistry as ‘‘boring, closed shell and all ionic, aqueous chem- istry, where everything is known! It is not worth investigating”. How wrong they are! Far from forming only purely ionic compounds, this review will shed light on some spectacular molecules, showing that alkaline earth metal ions can behave similar to transition metal ions. After general remarks about each element, from beryllium to barium, mainly recent examples of group 2 coordination compounds will be presented, their bonding situation will be discussed and examples of applications, e.g. in catalysis, will be given. Contents 1. Introduction . ........................................................................................................ 1 2. Beryllium . ........................................................................................................ 2 3. Magnesium . ........................................................................................................ 3 4. Calcium . ........................................................................................................ 7 5. Strontium . ....................................................................................................... 12 6. Barium . ...................................................................................................... -

Draft Toxicological Profile for Perchlorates
DRAFT TOXICOLOGICAL PROFILE FOR PERCHLORATES U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES Public Health Service Agency for Toxic Substances and Disease Registry September 2005 Additional Resources http://www.atsdr.cdc.gov/toxprofiles/tp162.html PERCHLORATES ii DISCLAIMER The use of company or product name(s) is for identification only and does not imply endorsement by the Agency for Toxic Substances and Disease Registry. This information is distributed solely for the purpose of pre dissemination public comment under applicable information quality guidelines. It has not been formally disseminated by the Agency for Toxic Substances and Disease Registry. It does not represent and should not be construed to represent any agency determination or policy. ***DRAFT FOR PUBLIC COMMENT*** PERCHLORATES iii UPDATE STATEMENT Toxicological profiles are revised and republished as necessary. For information regarding the update status of previously released profiles, contact ATSDR at: Agency for Toxic Substances and Disease Registry Division of Toxicology and Environmental Medicine/Toxicology Information Branch 1600 Clifton Road NE Mailstop F-32 Atlanta, Georgia 30333 ***DRAFT FOR PUBLIC COMMENT*** PERCHLORATES v FOREWORD This toxicological profile is prepared in accordance with guidelines developed by the Agency for Toxic Substances and Disease Registry (ATSDR) and the Environmental Protection Agency (EPA). The original guidelines were published in the Federal Register on April 17, 1987. Each profile will be revised and republished as necessary. The ATSDR toxicological profile succinctly characterizes the toxicologic and adverse health effects information for the hazardous substance described therein. Each peer-reviewed profile identifies and reviews the key literature that describes a hazardous substance’s toxicologic properties. -

Apparatus for Water Detection Using a Radioactive Tritium Labelled Reactant
United States Patent [is] 3,655,982 Gelezunas [45] Apr. II, 1972 [54] APPARATUS FOR WATER DETECTION OTHER PUBLICATIONS USING A RADIOACTIVE TRITIUM Holtman et al., " Radiometric Determination of Moisture in •Gases," Radioisotope Tracers in Industry and Geophysics, LABELLED REACTANT Symposium Proceedings Prague 1966, International Atomic [72] Inventor: Vincent L. Gelezunas, Devon, Pa. Agency, Vienna 1967, pp. 261-270. Thompson, R. C., Nucleonics, " Biological Applications of [73] Assignee: General Electric Company Tritium," Vol. 12,No. 9, Sept. 1954, pp. 31-35. [22] Filed: Jan. 29,1969 Primary Examiner—James W. Lawrence [21] Appl.No.: 795,111 Assistant Examiner—Davis L. Willis Attorney—William G. Becker, Paul F. Prestia, Allen E. Am- gott, Henry W. Kaufmann, Frank L. Neuhauser and Oscar B. [52] U.S. CI 250/106 T, 23/255 E, 250/43.5 MR, Waddell 250/83.6 FT [51] Int.Cl G21h 5/02 [57] ABSTRACT [58] Field of Search 250/106 T, 83.6 FT, 43.5 R; 23/232 C, 232 E, 255 E A method and apparatus for detecting water using an ap- propriate radioactive labelled alkaline earth hydride as a reac- tant. Fluid from a gas liquid or solid sample to be tested is [56] References Cited passed through a bed of the reactant. The water present in the UNITED STATES PATENTS sample reacts with the reactant forming a radioactive product whose radioactivity is then measured to give a quantitative 3,118,735 1/1964 Favreetal 23/230 determination of the amount of water in the sample or a qualitative determination of the presence of water. -

Danh Mục Số Cas A
CÔNG TY CỔ PHẦN CÔNG NGHIỆP VIỆT XUÂN NHÀ CUNG CẤP KHÍ HELI, KHÍ METAN, KHÍ SF6, KHÍ PROPAN Số 80B Nguyễn Văn Cừ - Long Biên - Hà Nội - Việt Nam www.vietxuangas.com. -
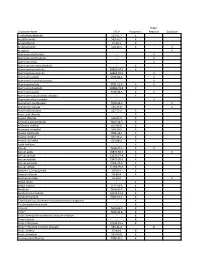
List of Major Physical Hazards
Water Substance Name CAS # Pyrophoric Reactive Explosive 2-ethylhexaldehyde 123-05-7 X Acetyl bromide 509-96-7 X Acetyl chloride 75-36-5 X Acetyl peroxide 110-22-5 X X Acetylene X Aluminum alkyl halides -- X Aluminum alkyl hydrides -- X Aluminum alkyls -- X Aluminum aminoborohydride -- X Aluminum borohydride 16962-07-5 X Aluminum borohydride 16962-07-5 X Aluminum carbide 1299-86-1 X Aluminum ferrosilcon powder -- X Aluminum hydride 7784-21-6 X X Aluminum phosphide 20859-73-8 X Aluminum powder 7429-90-5 X X Aluminum sesquibromide ethylate -- X Aluminum silicon powder -- X Ammonium perchlorate 7790-98-9 X Ammonium picrate 131-74-8 X Amyl trichlorosilane 107-72-2 X Anisic acid chloride -- X Anisoyl chloride 100-07-2 X Antimony pentachloride 7647-18-9 X Antimony, triethyl 617-85-6 X Antimony, trimethyl 594-10-5 X Arsenic trichloride 7784-34-1 X Arsenic, triethyl 617-75-4 X Arsenic, trimethyl 593-88-4 X Azido thallium -- X Barium 7440-39-3 X X Barium azide 18810-58-7 X X Barium carbide 12070-27-8 X Barium hydride 13477-09-3 X Barium peroxide 1304-29-6 X Barium sulfide 21109-95-5 X Benzene, 1,2-epoxyethyl 96-09-3 X Benzoyl chloride 98-88-4 X Benzoyl peroxide 94-36-0 X Benzyl silane -- X Benzyl sodium 1121-53-5 X Beryllium 7440-41-7 X Beryllium borohydride 30374-53-9 X Beryllium hydride 7787-52-2 X Bis(ethylamino) siloxenebis-cyclopentadienyl manganese -- X Bis-dimethylstibine oxide -- X Bismuth 7440-69-9 X Boron 7440-42-8 X Boron arsenotribromideboron chloride tetramer -- X Boron triazide -- X Boron tribromide 10294-33-4 X Boron trifluoride -

Ammonia Borane: an Extensively Studied, Though Not Yet Implemented, Hydrogen Carrier
energies Review Ammonia Borane: An Extensively Studied, Though Not Yet Implemented, Hydrogen Carrier Umit Bilge Demirci Institut Européen des Membranes, IEM—UMR 5635, ENSCM, CNRS, Univ Montpellier, F-34095 Montpellier, France; [email protected]; Tel.: +33-4-6714-9160 Received: 14 May 2020; Accepted: 11 June 2020; Published: 13 June 2020 Abstract: Ammonia borane H N BH (AB) was re-discovered, in the 2000s, to play an important role 3 − 3 in the developing hydrogen economy, but it has seemingly failed; at best it has lagged behind. The present review aims at analyzing, in the context of more than 300 articles, the reasons why AB gives a sense that it has failed as an anodic fuel, a liquid-state hydrogen carrier and a solid hydrogen carrier. The key issues AB faces and the key challenges ahead it has to address (i.e., those hindering its technological deployment) have been identified and itemized. The reality is that preventable errors have been made. First, some critical issues have been underestimated and thereby understudied, whereas others have been disproportionally considered. Second, the potential of AB has been overestimated, and there has been an undoubted lack of realistic and practical vision of it. Third, the competition in the field is severe, with more promising and cheaper hydrides in front of AB. Fourth, AB has been confined to lab benches, and consequently its technological readiness level has remained low. This is discussed in detail herein. Keywords: ammonia borane; dehydrocoupling; direct fuel cell; electro-oxidation; energy carrier; hydrogen carrier; hydrogen generation; hydrogen storage; hydrolysis; thermolysis 1. -

Maarten C. Verbraeken Phd Thesis
DOPED ALKALINE EARTH (NITRIDE) HYDRIDES Maarten Christiaan Verbraeken A Thesis Submitted for the Degree of PhD at the University of St. Andrews 2009 Full metadata for this item is available in the St Andrews Digital Research Repository at: https://research-repository.st-andrews.ac.uk/ Please use this identifier to cite or link to this item: http://hdl.handle.net/10023/714 This item is protected by original copyright Doped alkaline earth (nitride) hydrides A thesis presented for the degree of Doctor of Philosophy by Maarten Christiaan Verbraeken University of St Andrews Supervised by Prof. J.T.S. Irvine Submitted February 2009 Doped alkaline earth (nitride) hydrides Declarations I, Maarten Christiaan Verbraeken, hereby certify that this thesis, which is approximately 40,000 words in length, has been written by me, that it is the record of work carried out by me and that it has not been submitted in any previous application for a higher degree. I was admitted as a research student in June 2005 and as a candidate for the degree of Doctor of Philosophy in November 2006; the higher study for which this is a record was carried out in the University of St Andrews between 2005 and 2009. Date:……………….. Signature of candidate:………………………….. I hereby certify that the candidate has fulfilled the conditions of the Resolution and Regulations appropriate for the degree of Doctor of Philosophy in the University of St Andrews and that the candidate is qualified to submit this thesis in application for that degree. Date:……………….. Signature of supervisor:…………………………. In submitting this thesis to the University of St Andrews we understand that we are giving permission for it to be made available for use in accordance with the regulations of the University Library for the time being in force, subject to any copyright vested in the work not being affected thereby.