Organocatalytic Asymmetric Michael Addition of Ketones to Α, Β-Unsaturated Nitro Compounds
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Alkylation by Enamines for Synthesis of Some Heterocyclic Compounds ﻴﺩ
------J. Raf. Sci., Vol. 20, No.2, pp 92- 101, 2009 ------ Alkylation by Enamines for Synthesis of some Heterocyclic Compounds Jasim A. Abdullah Department of Chemistry College of Education Mosul University (Received 25/ 9/ 2008 ; Accepted 13 / 4 / 2009) ABSTRACT Compounds of 4-phenyl-3-butene-2-one (1) and 4-(4-chlorophenyl)-3-butene-2-one (2) were prepared by reaction of benzaldehyde or 4-chlorobenzaldehyde with acetone. Also 1,3-diphenyl-2-chloropropene-1-one (3) was synthesized from the reaction of benzaldehyde with 2-chloroacetophenone through Claisen-Shmidt condensation. The substituted ∆1(9)- octalone-2 (4,5) was prepared by reaction of the compounds (1and2) with cyclohexanone through Michael addition followed by aldol condensation. Compounds (4,5) were reacted with alkyl halide, as 2-chloroacetophenone or 1,3-diphenyl-2-chloropropene-1-one (3), via enamines formation which then hydrolyzed to give compounds (6-9). Compounds (6,7) were reacted with hydrazine , urea and thiourea to afford compounds (10-15) . The structures of all synthesized compounds were confirmed by available physical and spectral means . Keywords : Enamines , Heterocyclic compounds. ــــــــــــــــــــــــــــــــــــــــــــــــــ ﺍﻻﻟﻜﻠﺔ ﺒﻭﺍﺴﻁﺔ ﺍﻷﻴﻨﺎﻤﻴﻨﺎﺕ ﻟﺘﺸﻴﻴﺩ ﺒﻌﺽ ﺍﻟﻤﺭﻜﺒﺎﺕ ﺍﻟﺤﻠﻘﻴﺔ ﻏﻴﺭ ﺍﻟﻤﺘﺠﺎﻨﺴﺔ ﺍﻟﻤﻠﺨﺹ ﺘﻡ ﺘﺤﻀﻴﺭ ﺍﻟﻤﺭﻜﺒﺎﺕ 4-ﻓﻨﻴل-3-ﺒﻴﻭﺘﻴﻥ-2-ﺍﻭﻥ (1) ﻭ4-(4-ﻜﻠﻭﺭﻭﻓﻨﻴل)-3-ﺒﻴﻭﺘﻴﻥ-2-ﺍﻭﻥ (2) ﻤﻥ ﺘﻔﺎﻋل ﺍﻟﺒﻨﺯﺍﻟﺩﻴﻬﻴﺩ ﺃﻭ 4-ﻜﻠﻭﺭﻭﺍﻟﺒﻨﺯﺍﻟﺩﻴﻬﻴﺩ ﻤﻊ ﺍﻻﺴﻴﺘﻭﻥ . ﺤﻀﺭ ﺍﻟﻤﺭﻜﺏ 3,1-ﺜﻨﺎﺌﻲ ﻓﻨﻴل -2-ﻜﻠـﻭﺭﻭﺒﺭﻭﺒﻴﻥ -1-ﺍﻭﻥ (3) ﻤـﻥ ﺘﻔﺎﻋـل ﺍﻟﺒﻨﺯﺍﻟﺩﻴﻬﻴـﺩ ﻤـﻊ -2 9 1 ﻜﻠﻭﺭﻭﺍﺴﻴﺘﻭﻓﻴﻨﻭﻥ ﺒﻭﺴﺎﻁﺔ ﺘﻜﺎﺜﻑ (ﻜﻠﻴﺯﻥ-ﺸﻤﺩﺕ).ﺤﻀﺭﺕ ﻤﻌﻭﻀﺎﺕ ∆ ( ) –ﺍﻭﻜﺘﺎﻟﻭﻥ-2 ﻤﻥ ﺨﻼل ﺘﻔﺎﻋل ﺍﻟﻤﺭﻜﺒﻴﻥ (2,1) ﻤﻊ ﺍﻟﻬﻜﺴﺎﻨﻭﻥ ﺍﻟﺤﻠﻘﻲ ﺒﻭﺴﺎﻁﺔ ﺍﻀﺎﻓﺔ ﻤﺎﻴﻜل ﺍﻟﺘﻲ ﻴﺘﺒﻌﻬﺎ ﺘﻜﺎﺜﻑ ﺍﻻﻟﺩﻭل . -

Catalytic Direct Asymmetric Michael Reactions
ORGANIC LETTERS 2001 Catalytic Direct Asymmetric Michael Vol. 3, No. 23 Reactions: Taming Naked Aldehyde 3737-3740 Donors Juan M. Betancort and Carlos F. Barbas III* The Skaggs Institute for Chemical Biology and the Department of Molecular Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037 [email protected] Received September 5, 2001 ABSTRACT Direct catalytic enantio- and diastereoselective Michael addition reactions of unmodified aldehydes to nitro olefins using (S)-2-(morpholinomethyl)- pyrrolidine as a catalyst are described. The reactions proceed in good yield (up to 96%) in a highly syn-selective manner (up to 98:2) with enantioselectivities approaching 80%. The resulting γ-formyl nitro compounds are readily converted to chiral, nonracemic 3,4-disubstituted pyrrolidines. The Michael reaction is generally regarded as one of the Typically, carbon nucleophiles that contain an active most efficient carbon-carbon bond forming reactions, and methylene center such as malonic acid esters, â-keto esters, studies concerning this reaction have played an important nitroalkanes, etc. have been studied in the Michael reaction. role in the development of modern synthetic organic Carbonyl compounds, and ketones in particular, have gener- chemistry.1 As the demand for optically active compounds ally only been used as donors following their preactivation has soared in recent years, much progress has been made in by conversion into a more reactive species such as enol or the development of asymmetric variants of this reaction, enamine equivalents.5,6 In these cases, additional synthetic providing for the preparation of Michael adducts with high enantiomeric purity.2 Though remarkable advances have been (3) (a) Chataigner, I.; Gennari, C.; Ongeri, S.; Piarulli, U.; Ceccarelli, S. -

S41467-018-07534-X.Pdf
ARTICLE DOI: 10.1038/s41467-018-07534-x OPEN Differentiation between enamines and tautomerizable imines in the oxidation reaction with TEMPO Xiaoming Jie1, Yaping Shang1, Zhe-Ning Chen 1, Xiaofeng Zhang1, Wei Zhuang1 & Weiping Su1 Enamine and imine represent two of the most common reaction intermediates in syntheses, and the imine intermediates containing α-hydrogen often exhibit the similar reactivity to 1234567890():,; enamines due to their rapid tautomerization to enamine tautomers. Herein, we report that the minor structural difference between the enamine and the enamine tautomer derived from imine tautomerization results in the different chemo- and regioselectivity in the reaction of cyclohexanones, amines and TEMPO: the reaction of primary amines furnishes the formal oxygen 1,2-migration product, α-amino-enones, while the reaction of secondary amines under similar conditions generates exclusively arylamines via consecutive dehydrogenation on the cyclohexyl rings. The 18O-labeling experiment for α-amino-enone formation revealed that TEMPO served as oxygen transfer reagent. Experimental and computational studies of reaction mechanisms revealed that the difference in chemo- and regioselectivity could be ascribed to the flexible imine-enamine tautomerization of the imine intermediate con- taining an α-hydrogen. 1 State Key Laboratory of Structural Chemistry, Center for Excellence in Molecular Synthesis, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, 155 Yangqiao Road West, Fuzhou 350002, China. -

Recoverable Phospha-Michael Additions Catalyzed by a 4-N,N
molecules Article Recoverable Phospha-Michael Additions Catalyzed by a 4-N,N-Dimethylaminopyridinium Saccharinate Salt or a Fluorous Long-Chained Pyridine: Two Types of Reusable Base Catalysts Eskedar Tessema 1,†, Vijayanath Elakkat 1,† , Chiao-Fan Chiu 2,3,*, Jing-Hung Zheng 1, Ka Long Chan 1, Chia-Rui Shen 4,5, Peng Zhang 6,* and Norman Lu 1,7,* 1 Institute of Organic and Polymeric Materials, National Taipei University of Technology, Taipei 106, Taiwan; [email protected] (E.T.); [email protected] (V.E.); [email protected] (J.-H.Z.); [email protected] (K.L.C.) 2 Department of Pediatrics, Linkou Medical Center, Chang Gung Memorial Hospital, Taoyuan 333, Taiwan 3 Graduate Institute of Clinical Medical Sciences, College of Medicine, Chang Gung University, Taoyuan 333, Taiwan 4 Department of Medical Biotechnology and Laboratory Sciences, College of Medicine, Chang Gung University, Taoyuan 333, Taiwan; [email protected] 5 Department of Ophthalmology, Linkou Medical Center, Chang Gung Memorial Hospital, Taoyuan 333, Taiwan 6 Department of Chemistry, University of Cincinnati, Cincinnati, OH 45221-0172, USA 7 Development Center for Smart Textile, National Taipei University of Technology, Taipei 106, Taiwan Citation: Tessema, E.; Elakkat, V.; * Correspondence: [email protected] (C.-F.C.); [email protected] (P.Z.); Chiu, C.-F.; Zheng, J.-H.; Chan, K.L.; [email protected] (N.L.) Shen, C.-R.; Zhang, P.; Lu, N. † These authors contributed equally to this work. Recoverable Phospha-Michael Additions Catalyzed by a Abstract: Phospha-Michael addition, which is the addition reaction of a phosphorus-based nucle- 4-N,N-Dimethylaminopyridinium ophile to an acceptor-substituted unsaturated bond, certainly represents one of the most versatile and Saccharinate Salt or a Fluorous powerful tools for the formation of P-C bonds, since many different electrophiles and P nucleophiles Long-Chained Pyridine: Two Types can be combined with each other. -

Enamine-Based Organocatalysis with Proline And
Acc. Chem. Res. 2004, 37, 580-591 carbon bond in a stereospecific fashion. These same Enamine-Based Organocatalysis features are typically absent in traditional synthetic asym- with Proline and Diamines: The metric aldol methodologies.2,3 Our ideal synthetic catalyst would be one that could Development of Direct Catalytic generate enamines from any number of aldehydes or ketones and then direct bond-forming reactions with a Asymmetric Aldol, Mannich, wide variety of electrophiles beyond the carbonyl elec- Michael, and Diels-Alder trophiles of the aldol reaction. Further, since these cata- lysts also generate imines as part of their reaction mech- Reactions anism, electrophilic catalysis might facilitate diverse WOLFGANG NOTZ, FUJIE TANAKA, AND reactions with nucleophiles. Such an antibody might be termed an ªopen-active siteº catalytic antibody. Indeed, CARLOS F. BARBAS, III* The Skaggs Institute for Chemical Biology and the we were able to demonstrate that our aldolase antibodies Departments of Chemistry and Molecular Biology, The could catalyze not only a wide range of intra- and Scripps Research Institute, 10550 North Torrey Pines Road, intermolecular aldol reactions but also decarboxylation La Jolla, California 92037 reactions via imine catalysis and certain Michael reactions Received February 2, 2004 as well.2 Significantly for what would become our studies in organocatalysis, in our search for ªopen-active siteº ABSTRACT antibody catalysis, we studied the potential of aldolase Enamines and imines have long been recognized as key intermedi- antibodies to catalyze the addition of ketones to nitrosty- ates in enzyme catalysis, particularly within a class of enzymes renes in Michael reactions, the addition of ketones to organic chemists would very much like to emulate, the aldolases. -

Development of the Interrupted Nazarov Cyclization of Allenyl Vinyl Ketones, with Application to the Total Synthesis of the Cyclooctane Natural Product Roseadione
Development of the Interrupted Nazarov Cyclization of Allenyl Vinyl Ketones, with Application to the Total Synthesis of the Cyclooctane Natural Product Roseadione by Vanessa M. Marx Submitted in partial fulfilment of the requirements for the degree of Doctor of Philosophy at Dalhousie University Halifax, Nova Scotia May 2011 © Copyright by Vanessa M. Marx, 2011 DALHOUSIE UNIVERSITY DEPARTMENT OF CHEMISTRY The undersigned hereby certify that they have read and recommend to the Faculty of Graduate Studies for acceptance a thesis entitled “Development of the Interrupted Nazarov Cyclization of Allenyl Vinyl Ketones, with Application to the Total Synthesis of the Cyclooctane Natural Product Roseadione” by Vanessa M. Marx in partial fulfilment of the requirements for the degree of Doctor of Philosophy. Dated: May 19, 2011 Supervisor: _________________________________ Readers: _________________________________ _________________________________ _________________________________ Departmental Representative: _________________________________ ii DALHOUSIE UNIVERSITY DATE: May 19, 2011 AUTHOR: Vanessa M. Marx TITLE: Development of the Interrupted Nazarov Cyclization of Allenyl Vinyl Ketones, with Application to the Total Synthesis of the Cyclooctane Natural Product Roseadione DEPARTMENT OR SCHOOL: Department of Chemistry DEGREE: PhD CONVOCATION: October YEAR: 2011 Permission is herewith granted to Dalhousie University to circulate and to have copied for non-commercial purposes, at its discretion, the above title upon the request of individuals or institutions. I understand that my thesis will be electronically available to the public. The author reserves other publication rights, and neither the thesis nor extensive extracts from it may be printed or otherwise reproduced without the author’s written permission. The author attests that permission has been obtained for the use of any copyrighted material appearing in the thesis (other than the brief excerpts requiring only proper acknowledgement in scholarly writing), and that all such use is clearly acknowledged. -

Electron Deficient Heterocycle. Examples Abound
Pyridines Pyridines are the most commonly encountered π- electron deficient heterocycle. Examples abound. H O 4 N nifedipine 5 3 N NH2 (anti- angina) H CH3 6 2 MeO2C CO2Me N N 1 N NO2 nicotine niacin (nicitinamide) (in NADP) There are many classical syntheses of pyridines; only a few can be covered in the time available. 1. Hantzsch Synthesis Probably the conceptually simples pyridine synthesis would be a simple extension based on the Paal- Knorr pyrrole synthesis. In other words…. Paal-Knorr NH3 R5 R2 5 2 R N R OO H the shouldn't a 1,5-dicarbonyl do.... NH3 [O] 6 2 6 2 R R 6 2 R N R O O R N R H This is absolutely feasible. Furthermore, as you might expect, the 1,5-dicarbonyls are readily accessible by Michael reaction chemistry (1,4-, conjugate additions). The one- pot version of this, where the unsaturated carbonyl is prepared, the Michael reaction occurs, and the pyridine formation is accomplished, is quite common. It is called the Hantzsch synthesis. 4 O O 4 R O NH3 or O R O EtO2C CO2Et R4 NH4OAc EtO + + OEt EtO OEt ∆∆∆ H2O, O O O O OO ** R4 R4 FeCl3 EtO2C CO2Et EtO2C CO2Et ∆∆∆ H2O, N N H It’s rather forceful conditions, but the ester groups can be decarboxylated. The shown example is a symmetric case, but unsymmetrical ones can be done by doing the aldol condensation (here called a Knoevenagel) first, discretely, and then putting that compound (** ) into a separate reaction doing the subsequent steps. -

Approach and Synthesis of Strychnos Alkaloids
N° d'ordre : 4155 THÈSE Présentée à L'UNIVERSITÉ BORDEAUX I ÉCOLE DOCTORALE DES SCIENCES CHIMIQUES par Dawood Hosni DAWOOD POUR OBTENIR LE GRADE DE DOCTEUR SPÉCIALITÉ : CHIMIE ORGANIQUE ********************* TOWARDS THE SYNTHESIS OF MONOTERPENOIDS INDOLE ALKALOIDS OF THE ASPIDOSPERMATAN AND STRYCHNAN TYPE ********************* Soutenue le: 17 décembre 2010 Après avis de: MM. PIVA Olivier Professeur, Claude Bernard Lyon 1 Rapporteur PALE Patrick Professeur, Louis Pasteur Strasbourg 1 Rapporteur Devant la commission d'examen formée de : MM. PIVA Olivier Professeur, Claude Bernard Lyon 1 Rapporteur PALE Patrick Professeur, Louis Pasteur Strasbourg 1 Rapporteur POISSON Jean-François Chargé de recherche, CNRS Examinateur VINCENT Jean-Marc Directeur de recherche, CNRS Examinateur LANDAIS Yannick Professeur, Bordeaux 1 Directeur de thèse ROBERT Frédéric Chargé de recherche, CNRS Codirecteur de thèse - 2010 - Abbreviations ∆: reflux °C: celsius degrees Ac: acetyle ALB Aluminium Lithium bis(binaphthoxide) complex AIBN : azobis(isobutyronitrile) aq.: aqueous Ar : aromatic BINAP : 2,2'-bis(diphenylphosphino)-1,1'-binaphthyle BINAPO : 2-diphenylphosphino-2'-diphenylphosphinyl-1,1'-binaphthalene BINOL: 1,1’-bi-2-naphthol Boc: tert-butyloxycarbonyle BOX: Bisoxazoline Bz : benzoyle Bn: benzyle cat. : catalytic DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene DCM: dichloromethane DCC: dicyclohexacarbodiimide dr.: diastereomeric ratio DIBAL-H: diisobutylaluminium hydride DIPEA: diisopropyléthylamine (Hünig Base) DMAP: dimethylaminopyridine DME: dimethoxyethane -
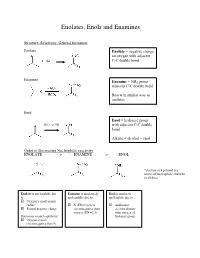
Enolates, Enols and Enamines
Enolates, Enols and Enamines Structure, Reactivity, General formation Enolate Enolate = negative charge on oxygen with adjacent C-C double bond Enamine Enamine = NR2 group adjacent C-C double bond Reacts in similar way as enolates Enol Enol = hydroxyl group - H3O+ or OH with adjacent C-C double bond Alkene + alcohol = enol Order of Decreasing Nucleophilic reactivity ENOLATE > ENAMINE > ENOL *electron-rich pi bond is a source of nucleophilic character in all three Enolate is nucleophilic due Enamine is moderately Enol is moderate to… nucleophilic due to… nucleophile due to… Oxygen’s small atomic radius N (EN=3) is less Additional Formal negative charge electronegative than electron density oxygen (EN =3.5) from oxygen of Detractors to nucleophilicity: hydroxyl group Oxygen is more electronegative than N When looking at which side is favored (products or reactants): Equilibrium favors side with WEAKER acid/base pair Weakest acid and weakest base should be on SAME side When comparing pKa values, juxtapose just the acids or just the bases Side with larger pka value (weaker acid) favored in equilibrium Remember that differences in pKa values reflect a difference quantity of 10x Example: compound H-A (pKa=3) + base compound A (pKa=10) + H-base 7 equilibrium lies to the RIGHT by factor of 10 Some Useful pKa values (from more acidic basic) H2SO4 (pka = -9) H2O (pka = 15.7) + H3O (pka = -1.8) CH3COCH3 (pka = 19) B-diketone (pka = 9) CH3COOCH3 (pKa = 25) HCN (pka = 9.1) LDA-H (pka = 36) CH3OH (pka = 15.5) How do we know which proton to deprotonate? Deprotonate most acidic proton usually leads to the most stable enolate 1. -

Advances in the Asymmetric Total Synthesis of Natural Products Using Chiral Secondary Amine Catalyzed Reactions of Α,Β-Unsaturated Aldehydes
molecules Review Advances in the Asymmetric Total Synthesis of Natural Products Using Chiral Secondary Amine Catalyzed Reactions of α,β-Unsaturated Aldehydes Zhonglei Wang 1,2 1 School of Pharmaceutical Sciences, Tsinghua University, Beijing 100084, China; [email protected] 2 School of Chemistry and Chemical Engineering, Qufu Normal University, Qufu 273165, China Academic Editor: Ari Koskinen Received: 12 August 2019; Accepted: 19 September 2019; Published: 19 September 2019 Abstract: Chirality is one of the most important attributes for its presence in a vast majority of bioactive natural products and pharmaceuticals. Asymmetric organocatalysis methods have emerged as a powerful methodology for the construction of highly enantioenriched structural skeletons of the target molecules. Due to their extensive application of organocatalysis in the total synthesis of bioactive molecules and some of them have been used in the industrial synthesis of drugs have attracted increasing interests from chemists. Among the chiral organocatalysts, chiral secondary amines (MacMillan’s catalyst and Jorgensen’s catalyst) have been especially considered attractive strategies because of their impressive efficiency. Herein, we outline advances in the asymmetric total synthesis of natural products and relevant drugs by using the strategy of chiral secondary amine catalyzed reactions of α,β-unsaturated aldehydes in the last eighteen years. Keywords: bioactive natural products; pharmaceuticals; asymmetric total synthesis; chiral secondary amines; α,β-unsaturated aldehydes 1. Introduction In 2000, the MacMillan group [1] first described the fundamental concept of LUMO-lowering iminium-ion activation. In 2005, the Jørgensen group [2,3] reported diarylprolinol silyl ether catalysts, which have also been used in iminium-ion catalysis. -

A Novel Aza-Nazarov Cyclization of Quinazolinonyl Enones: a Facile Access to C- Ring Substituted Vasicinones and Luotonins
A Novel Aza-Nazarov Cyclization of Quinazolinonyl Enones: A Facile Access to C- Ring Substituted Vasicinones and Luotonins Sivappa Rasapalli,a* Vamshikrishna Reddy Sammeta,a Zachary F. Murphy,a Yanchang Huang,a Jeffrey A. Boertha, James A. Golena and Sergey N. Savinovb a Department of Chemistry and Biochemistry, University of Massachusetts, 287 Old Westport Rd, N. Dartmouth, MA-02747, USA. b Department of Biochemistry and Molecular Biology. UMass Amherst, Amherst, MA-01003, USA [email protected] Received Date (will be automatically inserted after manuscript is accepted) ABSTRACT: A facile four-step synthetic access to C-ring substituted luotonins and vasicinones has been realized via a novel super acid mediated aza-Nazarov cyclization of quinazolinonyl enones. The regioselectivity of the cyclization is highly dependent on proton availability and overall polarity of the reaction medium. Quinazolinone is one of the most privileged through inhibition of topoisomerase-I (topo-I), through pharmacophores in medicinal chemistry.1, 2 There are mechanism of action analogous to camptothecin (CPT) more than 20 drugs that are currently in market (3),8 which attracted the attention of medchem and containing thequinazolinone core.3 Additionally, synchem communities alike.9 This increased attention is pharmacologically important natural products harboring partly due to toxic side effects associated with CPT and the quinazolinone core are also abundant and are known its analogs, due to hydrolysis of the E-ring in basic pH to possess wide range of -

The Abnormal Michael Reaction: Scope, Limitations, and Mechanism
Louisiana State University LSU Digital Commons LSU Historical Dissertations and Theses Graduate School 1971 The Abnormal Michael Reaction: Scope, Limitations, and Mechanism. William George Haag III Louisiana State University and Agricultural & Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_disstheses Recommended Citation Haag, William George III, "The Abnormal Michael Reaction: Scope, Limitations, and Mechanism." (1971). LSU Historical Dissertations and Theses. 2126. https://digitalcommons.lsu.edu/gradschool_disstheses/2126 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Historical Dissertations and Theses by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected]. 72-17,764 HAAG, 3rd., William George, 1946- THE ABNORMAL MICHAEL REACTION: SCOPE, LIMITATIONS, AND MECHANISM. The Louisiana State University and Agricultural and Mechanical College, Ph.D., 1971 Chemistry, organic University Microfilms, A XEROX Company, Ann Arbor, Michigan © 1972 WILLIAM GEORGE HAAG, 3rd. ALL RIGHTS RESERVED THE ABNORMAL MICHAEL REACTION: SCOPE, LIMITATIONS, AND MECHANISM A Dissertation Submitted to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College in partial fulfillment of the requirements for the degree of Doctor of Philosophy in The Department of Biochemistry by William George Haag, 3rd B.S., Louisiana State University, 1968 December 19JI PLEASE NOTE: Some pages may have indistinct print. Filmed as received. University Microfilms, A Xerox Education Company ACKNOWLEDGMENT The author would like to acknowledge Professor G.E. Risinger for his suggestions, discussion, and encouragement. His ever-present influence made completion of this paper possible.