RRM2 Is a Target for Synthetic Lethal Interactions with Replication Stress Checkpoint
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Phylogenetic Screening for Possible Novel
11 M060072591U NORTH-WEST UNIVERSITY tilt• YUNIBESITI YA BOKONE•BOPHIRIMA NOOROVVE S-UNIVERSITEIT PHYLOGENETIC SCREENING FOR POSSIBLE NOVEL ANTIBIOTIC PRODUCING ACTINOMYCETES FROM RHIZOSPHERIC SOIL SAMPLES COLLECTED FROM NGAKA MODIRI MOLEMA DISTRICT IN NORTH WEST PROVINCE, SOUTH AFRICA I BY MOBOLAJI FELICIA ADEGBOYE A thesis submitted in fulfilment of the requirements for the degree of DOCTOR OF PHILOSOPHY (BIOLOGY) DEPARTMENT OF BIOLOGICAL SCIENCES FACULTY OF SCIENCE, AGRICULTURE AND TECHNOLOGY NORTH-WEST UNIVERSITY, MAFIKENG CAMPUS SOUTH AFRICA Supervisor: Professor Olubukola 0. Babalola 2014 LIBRARY o MAFIKENG CAMPUS CALL NO.: 2021 -02- 0 4 DECLARATION I, the undersigned, declare that this thesis submitted to the North-West University for the degree of Doctor of Philosophy in Biology in the Faculty of Science, Agriculture and Technology, School of Environmental and Health Sciences, and the work contained herein is my original work with exemption to the citations and that this work has not been submitted at any other University in partial or entirely for the award of any degree. Name: Mobolaji Felicia Adegboye Signature: .....~ •·· ··· ····· ·· .. ··············· ..... Date: .... ~S.. .. ....a~ ·1·· ·'.}Q~i; ... ............ .... DEDICATION This work is dedicated to Almighty God for His faithfulness over my life and for making my helpers to be many. ii ACKNOWLEDGEMENTS I would like to express my deepest thanks, gratitude and appreciation to my supervisor and mentor, Prof. Olubukola 0. Babalola for giving me the opportunity to pursue my doctoral degree under her supervision and for her encouragement, help and kind support. Her invaluable advice, suggestions, discussions and guidance were a real support to me. I acknowledge with honour and gratitude the International Foundation for Science (IFS) for research grant (F/5330-1 ), Connect Africa Scholarship Award, H3ABioNet/SANBio Scholarship and North-West University for offering me bursary/scholarship award to pursue the PhD degree. -

Exosomes Confer Chemoresistance to Pancreatic Cancer Cells By
FULL PAPER British Journal of Cancer (2017) 116, 609–619 | doi: 10.1038/bjc.2017.18 Keywords: chemoresistance; exosomes; pancreatic cancer; ROS; microRNA Exosomes confer chemoresistance to pancreatic cancer cells by promoting ROS detoxification and miR-155-mediated suppression of key gemcitabine-metabolising enzyme, DCK Girijesh Kumar Patel1, Mohammad Aslam Khan1, Arun Bhardwaj1, Sanjeev K Srivastava1, Haseeb Zubair1, Mary C Patton1, Seema Singh1,2, Moh’d Khushman3 and Ajay P Singh*,1,2 1Department of Oncologic Sciences, Mitchell Cancer Institute, University of South Alabama, Mobile, AL, USA; 2Department of Biochemistry and Molecular Biology, College of Medicine, University of South Alabama, Mobile, AL, USA and 3Department of Interdisciplinary Clinical Oncology, Mitchell Cancer Institute, University of South Alabama, Mobile, AL, USA Background: Chemoresistance is a significant clinical problem in pancreatic cancer (PC) and underlying molecular mechanisms still remain to be completely understood. Here we report a novel exosome-mediated mechanism of drug-induced acquired chemoresistance in PC cells. Methods: Differential ultracentrifugation was performed to isolate extracellular vesicles (EVs) based on their size from vehicle- or gemcitabine-treated PC cells. Extracellular vesicles size and subtypes were determined by dynamic light scattering and marker profiling, respectively. Gene expression was examined by qRT-PCR and/or immunoblot analyses, and direct targeting of DCK by miR-155 was confirmed by dual-luciferase 30-UTR reporter assay. Flow cytometry was performed to examine the apoptosis indices and reactive oxygen species (ROS) levels in PC cells using specific dyes. Cell viability was determined using the WST-1 assay. Results: Conditioned media (CM) from gemcitabine-treated PC cells (Gem-CM) provided significant chemoprotection to subsequent gemcitabine toxicity and most of the chemoresistance conferred by Gem-CM resulted from its EVs fraction. -

Integrative Systems Biology– Renal Diseases: a Road to a Holist View of Chronic Disease Mechanism
Integrative Systems biology– Renal Diseases: A road to a holist view of chronic disease mechanism Matthias Kretzler Div. Nephrology / Internal Medicine Computational Medicine and Bioinformatics University of Michigan Medical School The challenge in chronic disease • Descriptive disease categorization with multiple pathogenetic mechanisms § Problems of ‘mixed bag’ diseases: • Unpredictable disease course and response to therapy • Nephrology as an ‘art of trial and error’ • Shift in our disease paradigms: § Mechanism based patient management • Define the disease process active in the individual patient – Base prognosis on specific disease process – Target therapy to interfere with the mechanism currently destroying endorgan function Molecular Nephrology approach Clinical outcome research Genetics Molecular Pathology Molecular Epigenetics Phenotyping Genomics Functional Clinical research Disease Genomics Biobanks Proteomics Model systems Animal models Molecular interaction In vitro tissue culture model systems Organ culture and development Tower of Babylon: Search for the universal language for the medicine of the 21st century Pieter Bruegl: 1563. Kunsthistorisches Museum Wien Molecular Nephrology approach Clinical outcome research Genetics Molecular Pathology Molecular Epigenetics Phenotyping Genomics Functional Clinical research Disease Genomics Integrative Biobanks Proteomics Biology (Physiology) Model systems Animal models Molecular interaction of renal disease In vitro tissue culture model systems Organ culture and development Systems -

Tremblay Robinlee.Pdf
Expression and Characterisation ofa Gene Enc oding RbpD, an RNA- Bind ing Protein in Anabaena sp. strain PeC 7120 by Rob in lee Tremblay A lhesis submitted to the Scltool of Graduale Studies in partial fulfilment of the requirements fOl" the degree of Master of Science Department of BiochemistrylFacultyof Science Memorial University of Newfoundland January 2000 SI.JOM'S Newfoun dland Abs t ra ct The RNA-binding protein RbpD, from the cyano bacterium Anaba ena sp, strain Pe C 7120 was expressed in £Sch~ ric h ia coli and successfully purified using me IMPACT I system (New England Biolabs). The rbp D gene was cloned into the pCYBt expre ssion vector by using polymerase chain reaction to introduce Ndel and SapI restriction sites at the 5' end 3' ends of the gene respect ively. The 3'.-end mutagenesis also chan ged the stop codon into a cysteine codon. The resulting gene encoded a fusion protein consisting of RbpD, the Saccharomyces cerev isiae VMA intein and a chitin binding domain.. Expressi on of the fusion protein was observed in £ coli strain MCI061 but Western blot analysis using an intein-directed ant ibody indicated that significant in vivo fmeln-direcred splicing of the fusion protein occurred. We were unable to eliminate this problem; no fusion protein expression was observed in 8 other E coli strains tested. Wild -type RbpD was purified following binding of the fusion protein 10 a chitin column and overnight cleavage in the presence of a reducing agent, dlthicthrehc l. A number of modifications to the manufacturer' s purification protocol were found to be necessary for success ful purification. -

Purification and Characterisation of a Protease (Tamarillin) from Tamarillo Fruit
Purification and characterisation of a protease (tamarillin) from tamarillo fruit Item Type Article Authors Li, Zhao; Scott, Ken; Hemar, Yacine; Zhang, Huoming; Otter, Don Citation Li Z, Scott K, Hemar Y, Zhang H, Otter D (2018) Purification and characterisation of a protease (tamarillin) from tamarillo fruit. Food Chemistry. Available: http://dx.doi.org/10.1016/ j.foodchem.2018.02.091. Eprint version Post-print DOI 10.1016/j.foodchem.2018.02.091 Publisher Elsevier BV Journal Food Chemistry Rights NOTICE: this is the author’s version of a work that was accepted for publication in Food Chemistry. Changes resulting from the publishing process, such as peer review, editing, corrections, structural formatting, and other quality control mechanisms may not be reflected in this document. Changes may have been made to this work since it was submitted for publication. A definitive version was subsequently published in Food Chemistry, [, , (2018-02-16)] DOI: 10.1016/j.foodchem.2018.02.091 . © 2018. This manuscript version is made available under the CC-BY-NC-ND 4.0 license http://creativecommons.org/licenses/by-nc-nd/4.0/ Download date 29/09/2021 23:19:14 Link to Item http://hdl.handle.net/10754/627180 Accepted Manuscript Purification and characterisation of a protease (tamarillin) from tamarillo fruit Zhao Li, Ken Scott, Yacine Hemar, Huoming Zhang, Don Otter PII: S0308-8146(18)30327-3 DOI: https://doi.org/10.1016/j.foodchem.2018.02.091 Reference: FOCH 22475 To appear in: Food Chemistry Received Date: 25 October 2017 Revised Date: 13 February 2018 Accepted Date: 16 February 2018 Please cite this article as: Li, Z., Scott, K., Hemar, Y., Zhang, H., Otter, D., Purification and characterisation of a protease (tamarillin) from tamarillo fruit, Food Chemistry (2018), doi: https://doi.org/10.1016/j.foodchem. -

Anticancer Potential of Resveratrol, -Lapachone and Their Analogues
molecules Review Anticancer Potential of Resveratrol, β-Lapachone and Their Analogues 1, 2, Danielly C. Ferraz da Costa y, Luciana Pereira Rangel y , 3, 3, Mafalda Maria Duarte da Cunha Martins-Dinis y, Giulia Diniz da Silva Ferretti y, Vitor F. Ferreira 4 and Jerson L. Silva 3,* 1 Departamento de Nutrição Básica e Experimental, Instituto de Nutrição, Universidade do Estado do Rio de Janeiro, Rio de Janeiro 20550-013, Brazil; [email protected] 2 Faculdade de Farmácia, Universidade Federal do Rio de Janeiro, Rio de Janeiro 21941-902, Brazil; [email protected] 3 Programa de Biologia Estrutural, Instituto de Bioquímica Médica Leopoldo de Meis, Instituto Nacional de Ciência e Tecnologia de Biologia Estrutural e Bioimagem, Universidade Federal do Rio de Janeiro, Rio de Janeiro 21941-902, Brazil; [email protected] (M.M.D.d.C.M.-D.); [email protected] (G.D.d.S.F.) 4 Departamento de Tecnologia Farmacêutica, Faculdade de Farmácia, Universidade Federal Fluminense, Rio de Janeiro 24241-000, Brazil; [email protected]ff.br * Correspondence: [email protected] These authors contributed equally to this work. y Received: 31 December 2019; Accepted: 13 February 2020; Published: 18 February 2020 Abstract: This review aims to explore the potential of resveratrol, a polyphenol stilbene, and beta-lapachone, a naphthoquinone, as well as their derivatives, in the development of new drug candidates for cancer. A brief history of these compounds is reviewed along with their potential effects and mechanisms of action and the most recent attempts to improve their bioavailability and potency against different types of cancer. Keywords: resveratrol; β-lapachone; cancer 1. -

Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degenerations: Similarities in Genetic Background
diagnostics Review Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degenerations: Similarities in Genetic Background Eva Parobkova 1 and Radoslav Matej 1,2,3,* 1 Department of Pathology and Molecular Medicine, Third Faculty of Medicine, Charles University and Thomayer University Hospital, 14059 Prague, Czech Republic; [email protected] 2 Department of Pathology, First Faculty of Medicine, Charles University, and General University Hospital, 14059 Prague, Czech Republic 3 Department of Pathology, Third Faculty of Medicine, Charles University, and University Hospital Kralovske Vinohrady, 14059 Prague, Czech Republic * Correspondence: [email protected] Abstract: Amyotrophic lateral sclerosis (ALS) is a devastating, uniformly lethal progressive degen- erative disorder of motor neurons that overlaps with frontotemporal lobar degeneration (FTLD) clinically, morphologically, and genetically. Although many distinct mutations in various genes are known to cause amyotrophic lateral sclerosis, it remains poorly understood how they selectively impact motor neuron biology and whether they converge on common pathways to cause neuronal degeneration. Many of the gene mutations are in proteins that share similar functions. They can be grouped into those associated with cell axon dynamics and those associated with cellular phagocytic machinery, namely protein aggregation and metabolism, apoptosis, and intracellular nucleic acid transport. Analysis of pathways implicated by mutant ALS genes has provided new insights into the pathogenesis of both familial forms of ALS (fALS) and sporadic forms (sALS), although, regrettably, this has not yet yielded definitive treatments. Many genes play an important role, with TARDBP, Citation: Parobkova, E.; Matej, R. SQSTM1, VCP, FUS, TBK1, CHCHD10, and most importantly, C9orf72 being critical genetic players Amyotrophic Lateral Sclerosis and in these neurological disorders. -

Nucleotide Biosynthesis Links Glutathione Metabolism to Ferroptosis Sensitivity
bioRxiv preprint doi: https://doi.org/10.1101/2021.07.14.452394; this version posted July 15, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Nucleotide Biosynthesis Links Glutathione Metabolism to Ferroptosis Sensitivity Amy Tarangelo1,3, Joon Tae Kim2, Jonathan Z. Long2, Scott J. Dixon1,4 1Department of Biology, Stanford University, 327 Campus Drive, Stanford, CA 94305, USA 2Department of Pathology and Stanford ChEM-H, Stanford University School of Medicine 291 Campus Drive, Stanford, CA 94305, USA 3Current address: Children’s Medical Center Research Institute, UT Southwestern, 6000 Harry Hines Blvd, Dallas, TX 75235 4Contact: [email protected] Key words: Cell death, iron, p53, p21 Running title: Nucleotide metabolism and ferroptosis bioRxiv preprint doi: https://doi.org/10.1101/2021.07.14.452394; this version posted July 15, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract Nucleotide synthesis is a metabolically demanding process essential for cell division. Several anti-cancer drugs that inhibit nucleotide metabolism induce apoptosis. How inhibition of nucleotide metabolism impacts non-apoptotic cell death is less clear. Here, we report that inhibition of nucleotide metabolism by the p53 pathway is sufficient to suppress the non-apoptotic cell death process of ferroptosis. -

Gene Metabolic Signature for Survival Prediction in Patients with Kidney Renal Clear Cell Carcinoma
www.aging-us.com AGING 2021, Vol. 13, No. 6 Research Paper Identification and validation of a two-gene metabolic signature for survival prediction in patients with kidney renal clear cell carcinoma Xudong Guo1, Zhuolun Sun2, Shaobo Jiang1, Xunbo Jin1, Hanbo Wang1 1Department of Urology, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan 250021, Shandong, China 2Department of Urology, Third Affiliated Hospital of Sun Yat-sen University, Guangzhou 510630, Guangdong, China Correspondence to: Hanbo Wang; email: [email protected], https://orcid.org/0000-0002-6095-8611 Keywords: kidney renal clear cell carcinoma, metabolism, nomogram, prognostic signature, The Cancer Genome Atlas Received: September 2, 2020 Accepted: October 22, 2020 Published: March 03, 2021 Copyright: © 2021 Guo et al. This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Metabolic reprogramming contributes to the high mortality of advanced stage kidney renal clear cell carcinoma (KIRC), the most common renal cancer subtype. This study aimed to identify a metabolism-related gene (MRG) signature to improve survival prediction in KIRC patients. We downloaded RNA sequencing data and corresponding clinical information for KIRC and control samples from The Cancer Genome Atlas database and identified, based on an MRG dataset in the Molecular Signatures Database, 123 MRGs with differential expression in KIRC. Following Cox regression analysis and least absolute shrinkage and selection operator selection, RRM2 and ALDH6A1 were identified as prognosis-related genes and used to construct a prognostic signature with independent prognostic significance. -
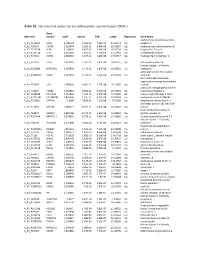
Table S1. the Statistical Metrics for Key Differentially Expressed Genes (Degs)
Table S1. The statistical metrics for key differentially expressed genes (DEGs) Gene Agilent Id Symbol logFC pValue FDR tvalue Regulation Gene Name oxidized low density lipoprotein A_24_P124624 OLR1 2.458429 1.19E-13 7.25E-10 24.04241 Up receptor 1 A_23_P90273 CHST8 2.622464 3.85E-12 6.96E-09 19.05867 Up carbohydrate sulfotransferase 8 A_23_P217528 KLF8 2.109007 4.85E-12 7.64E-09 18.76234 Up Kruppel like factor 8 A_23_P114740 CFH 2.651636 1.85E-11 1.79E-08 17.13652 Up complement factor H A_23_P34031 XAGE2 2.000935 2.04E-11 1.81E-08 17.02457 Up X antigen family member 2 A_23_P27332 TCF4 1.613097 2.32E-11 1.87E-08 16.87275 Up transcription factor 4 histone cluster 1 H1 family A_23_P250385 HIST1H1B 2.298658 2.47E-11 1.87E-08 16.80362 Up member b abnormal spindle microtubule A_33_P3288159 ASPM 2.162032 2.79E-11 2.01E-08 16.66292 Up assembly H19, imprinted maternally expressed transcript (non-protein A_24_P52697 H19 1.499364 4.09E-11 2.76E-08 16.23387 Up coding) potassium voltage-gated channel A_24_P31627 KCNB1 2.289689 6.65E-11 3.97E-08 15.70253 Up subfamily B member 1 A_23_P214168 COL12A1 2.155835 7.59E-11 4.15E-08 15.56005 Up collagen type XII alpha 1 chain A_33_P3271341 LOC388282 2.859496 7.61E-11 4.15E-08 15.55704 Up uncharacterized LOC388282 A_32_P150891 DIAPH3 2.2068 7.83E-11 4.22E-08 15.5268 Up diaphanous related formin 3 zinc finger protein 185 with LIM A_23_P11025 ZNF185 1.385721 8.74E-11 4.59E-08 15.41041 Up domain heat shock protein family B A_23_P96872 HSPB11 1.887166 8.94E-11 4.64E-08 15.38599 Up (small) member 11 A_23_P107454 -

Novel Insights Into the Molecular Regulation of Ribonucleotide Reductase in Adrenocortical Carcinoma Treatment
cancers Article Novel Insights into the Molecular Regulation of Ribonucleotide Reductase in Adrenocortical Carcinoma Treatment Christina Bothou 1,2,† , Ashish Sharma 1,†, Adrian Oo 3, Baek Kim 3,4, Pal Perge 5 , Peter Igaz 5,6 , Cristina L. Ronchi 7,8,9, Igor Shapiro 1 and Constanze Hantel 1,10,* 1 Department of Endocrinology, Diabetology and Clinical Nutrition, University Hospital Zurich (USZ), University of Zurich (UZH), CH-8091 Zurich, Switzerland; [email protected] (C.B.); [email protected] (A.S.); [email protected] (I.S.) 2 Competence Centre of Personalized Medicine, Molecular and Translational Biomedicine PhD Program, University of Zurich (UZH), CH-8006 Zurich, Switzerland 3 Department of Pediatrics, School of Medicine, Emory University, Atlanta, GA 30322, USA; [email protected] (A.O.); [email protected] (B.K.) 4 Center for Drug Discovery, Children’s Healthcare of Atlanta, Atlanta, GA 30322, USA 5 Department of Endocrinology, Department of Internal Medicine and Oncology, Faculty of Medicine, Semmelweis University, H-1083 Budapest, Hungary; [email protected] (P.P.); [email protected] (P.I.) 6 MTA-SE Molecular Medicine Research Group, H-1083 Budapest, Hungary 7 Division of Endocrinology and Diabetes, Department of Medicine I, University Hospital of Wuerzburg, University of Wuerzburg, 97080 Wuerzburg, Germany; [email protected] 8 Institute of Metabolism and Systems Research, University of Birmingham, Birmingham B15 2TT, UK 9 Centre for Endocrinology, Diabetes and Metabolism, Birmingham Health Partners, Birmingham B15 2TT, UK 10 Medizinische Klinik und Poliklinik III, University Hospital Carl Gustav Carus Dresden, 01307 Dresden, Germany Citation: Bothou, C.; Sharma, A.; Oo, * Correspondence: [email protected]; Tel.: +41-43-253-3008 A.; Kim, B.; Perge, P.; Igaz, P.; Ronchi, † Equally contributing first authors. -

PLEK2, RRM2, GCSH: a Novel WWOX-Dependent Biomarker Triad of Glioblastoma at the Crossroads of Cytoskeleton Reorganization and Metabolism Alterations
cancers Article PLEK2, RRM2, GCSH: A Novel WWOX-Dependent Biomarker Triad of Glioblastoma at the Crossroads of Cytoskeleton Reorganization and Metabolism Alterations Zaneta˙ Kałuzi ´nska* , Damian Kołat , Andrzej K. Bednarek and Elzbieta˙ Płuciennik Department of Molecular Carcinogenesis, Medical University of Lodz, 90-752 Lodz, Poland; [email protected] (D.K.); [email protected] (A.K.B.); [email protected] (E.P.) * Correspondence: [email protected] Simple Summary: Cytoskeleton reorganization affects the malignancy of glioblastoma. The WWOX gene is a tumor suppressor in glioblastoma and was found to modulate the cytoskeletal machinery in neural progenitor cells. To date, the role of this gene in the cytoskeleton or glioblastoma has been studied separately. Therefore, the purpose of this study was to investigate WWOX-dependent genes in glioblastoma and indicate cytoskeleton-related processes they are involved in. The most relevant WWOX-dependent genes were found to be PLEK2, RRM2, and GCSH, which have been proposed as novel biomarkers. Their biological functions suggest that there is an important link between cytoskeleton and metabolism, orchestrating tumor proliferation, metastasis, and resistance. Searching for such new therapeutic targets is important due to the constant lack of effective treatment ˙ Citation: Kałuzi´nska, Z.; Kołat, D.; for glioblastoma patients. Bednarek, A.K.; Płuciennik, E. PLEK2, RRM2, GCSH: A Novel Abstract: Glioblastoma is one of the deadliest human cancers. Its malignancy depends on cytoskele- WWOX-Dependent Biomarker Triad ton reorganization, which is related to, e.g., epithelial-to-mesenchymal transition and metastasis. The of Glioblastoma at the Crossroads of malignant phenotype of glioblastoma is also affected by the WWOX gene, which is lost in nearly Cytoskeleton Reorganization and a quarter of gliomas.