Epithelial Ablation of Miro1/Rhot1 Gtpase Leads to Mitochondrial
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
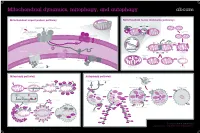
Mitochondrial Dynamics, Mitophagy, and Autophagy
Mitochondrial dynamics, mitophagy, and autophagy Mitochondrial import protein pathway Mitochondrial fusion and fission pathways iΔΨm, (OXPHOSh) Ca2+ Fusion ++++ (Cell differentiationi) N Internal signal β-barrel outer Opa1 SH ER sequence SH membrane precursors Mfn1/2 SH SH GTP GTP C Inner membrane and GTP GTP Outer membrane Matrix carrier precursors 22 70 GTP fusion (N-terminal presequences) 35 GTP 20 37 Tom complex 5 Sam complex Opa1 Cytosol Outer 6 Sam50 7 Mdm10 membrane DRP1 Fis1 Inner membrane fusion Tim8-Tim13 Mim 1 Tom40 Bax/Bak apoptosis Outer membrane HR2 regions S-S Tim9-Tim10 S-S Inner Erv1 95 A 54 mitochondrial S-S Reorganization Mia40 space 21 Fission sequestration 50 18 Tim22 Translocation Depolarization to meet iΔΨm ATP needs Tim23 KREBS Cycle NADH FADH2 17 Oxa1 Tim 22 complex Mia Mba1 Mdm I ATP 44 38 H+ II 17 H+ Inner membrane 16 +++ III ADP - Pi 18 Ribosome Oxa H+ IV mtHsp70 H+ ATP +++ Mge1 MPP Healthy mitochondrion Mitophagy Matrix Tim 23 complex Mitophagy pathway Autophagy pathway Lysosomal hydroiase Lysosome Hypoxia, Nutrient/Growth Rapamycin Starvation condition Ubiquitin Cytosol Parkin factor deprivation Lamp Stress Damaged mitochondrion Parkin Parkin iΔΨm Parkin MARF PINK1 PINK1 PINK1 Mfn1 VDAC1 Parkin AMPK mTOR Mfn2 PINK1 PINK1 Phagophore Autophagosome Autophagolyosome PARL LC3-II PINK1-L PINK1-S Parkin P FIS1 ATG13 ULK LC3-II Membrane PINK1 PINK1 FIP200 PINK1 MIRO2 P Bak PINK1 MIRO1 Parkin Parkin Parkin Cytoplasmic Cytosol proLC3 macromolecule LC3-II Atg4B Atg7/LC3-I Atg5/Atg12/Atg16 Organelle Atg9 PE Atg18 Atg2 -

PARSANA-DISSERTATION-2020.Pdf
DECIPHERING TRANSCRIPTIONAL PATTERNS OF GENE REGULATION: A COMPUTATIONAL APPROACH by Princy Parsana A dissertation submitted to The Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland July, 2020 © 2020 Princy Parsana All rights reserved Abstract With rapid advancements in sequencing technology, we now have the ability to sequence the entire human genome, and to quantify expression of tens of thousands of genes from hundreds of individuals. This provides an extraordinary opportunity to learn phenotype relevant genomic patterns that can improve our understanding of molecular and cellular processes underlying a trait. The high dimensional nature of genomic data presents a range of computational and statistical challenges. This dissertation presents a compilation of projects that were driven by the motivation to efficiently capture gene regulatory patterns in the human transcriptome, while addressing statistical and computational challenges that accompany this data. We attempt to address two major difficulties in this domain: a) artifacts and noise in transcriptomic data, andb) limited statistical power. First, we present our work on investigating the effect of artifactual variation in gene expression data and its impact on trans-eQTL discovery. Here we performed an in-depth analysis of diverse pre-recorded covariates and latent confounders to understand their contribution to heterogeneity in gene expression measurements. Next, we discovered 673 trans-eQTLs across 16 human tissues using v6 data from the Genotype Tissue Expression (GTEx) project. Finally, we characterized two trait-associated trans-eQTLs; one in Skeletal Muscle and another in Thyroid. Second, we present a principal component based residualization method to correct gene expression measurements prior to reconstruction of co-expression networks. -

Amyloid Β-Peptide Increases Mitochondria-Endoplasmic
cells Article Amyloid β-Peptide Increases Mitochondria-Endoplasmic Reticulum Contact Altering Mitochondrial Function and Autophagosome Formation in Alzheimer’s Disease-Related Models Nuno Santos Leal 1,*, Giacomo Dentoni 1 , Bernadette Schreiner 1 , Luana Naia 1, Antonio Piras 1, Caroline Graff 1, Antonio Cattaneo 2, Giovanni Meli 2 , Maho Hamasaki 3, Per Nilsson 1 and Maria Ankarcrona 1,* 1 Division of Neurogeriatrics, Department of Neurobiology, Care Science and Society, Karolinska Institutet, BioClinicum J9:20, Visionsgatan 4, 171 64 Solna, Sweden; [email protected] (G.D.); [email protected] (B.S.); [email protected] (L.N.); [email protected] (A.P.); caroline.graff@ki.se (C.G.); [email protected] (P.N.) 2 European Brain Research Institute (EBRI), Viale Regina Elena 295, 00161 Roma, Italy; [email protected] (A.C.); [email protected] (G.M.) 3 Department of Genetics, Graduate School of Medicine, Osaka University, 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan; [email protected] * Correspondence: [email protected] (N.S.L.); [email protected] (M.A.); Tel.: +44-122-333-4390 (N.S.L.); +46-852-483-577 (M.A.) Received: 23 October 2020; Accepted: 25 November 2020; Published: 28 November 2020 Abstract: Recent findings have shown that the connectivity and crosstalk between mitochondria and the endoplasmic reticulum (ER) at mitochondria–ER contact sites (MERCS) are altered in Alzheimer’s disease (AD) and in AD-related models. MERCS have been related to the initial steps of autophagosome formation as well as regulation of mitochondrial function. Here, the interplay between MERCS, mitochondria ultrastructure and function and autophagy were evaluated in different AD animal models with increased levels of Aβ as well as in primary neurons derived from these animals. -

Involvement of Impaired Autophagy and Mitophagy in Neuro-2A Cell
www.nature.com/scientificreports OPEN Involvement of impaired autophagy and mitophagy in Neuro-2a cell damage under hypoxic and/or high- Received: 19 May 2017 Accepted: 15 January 2018 glucose conditions Published: xx xx xxxx Yufei Song, Yu Du, Wenying Zou, Yan Luo, Xiaojie Zhang & Jianliang Fu Chronic cerebral hypoperfusion (CCH) plays an insidious role in the development of cognitive impairment. Considerable evidence suggests that Diabetes Mellitus (DM) as a vascular risk factor may exacerbate CCH and is closely related to cognitive decline. Dysregulation of autophagy is known to be associated with the pathogenesis of neurodegenerative diseases such as Alzheimer’s disease. To elucidate the role of autophagy in CCH- and/or DM-related pathogenesis, mouse neuroblastoma Neuro-2a cells were exposed to hypoxia and/or high glucose for 48 h, mimicking CCH complicated with DM pathologies. Chronic hypoxia reduced cell proliferation and increased levels of cleaved caspase-3, whereas high glucose had no obvious synergistic toxic efect. Accumulation of autophagic vacuoles under hypoxia may be due to both autophagy impairment and induction, with the former accounting for Neuro-2a cell death. Additionally, aberrant accumulation of mitochondria in Neuro-2a cells may be attributed to insufcient BNIP3-mediated mitophagy due to poor interaction between BNIP3 and LC3-II. Despite the lack of a signifcant cytotoxic efect of high glucose under our experimental conditions, our data indicated for the frst time that impaired autophagy degradation and inefcient BNIP3-mediated mitophagy may constitute mechanisms underlying neuronal cell damage during chronic hypoxia. Chronic cerebral hypoperfusion (CCH) is a normal process related to ageing that likely contributes to age-related memory loss1. -

The Mitochondrial Kinase PINK1 in Diabetic Kidney Disease
International Journal of Molecular Sciences Review The Mitochondrial Kinase PINK1 in Diabetic Kidney Disease Chunling Huang * , Ji Bian , Qinghua Cao, Xin-Ming Chen and Carol A. Pollock * Kolling Institute, Sydney Medical School, Royal North Shore Hospital, University of Sydney, St. Leonards, NSW 2065, Australia; [email protected] (J.B.); [email protected] (Q.C.); [email protected] (X.-M.C.) * Correspondence: [email protected] (C.H.); [email protected] (C.A.P.); Tel.: +61-2-9926-4784 (C.H.); +61-2-9926-4652 (C.A.P.) Abstract: Mitochondria are critical organelles that play a key role in cellular metabolism, survival, and homeostasis. Mitochondrial dysfunction has been implicated in the pathogenesis of diabetic kidney disease. The function of mitochondria is critically regulated by several mitochondrial protein kinases, including the phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1). The focus of PINK1 research has been centered on neuronal diseases. Recent studies have revealed a close link between PINK1 and many other diseases including kidney diseases. This review will provide a concise summary of PINK1 and its regulation of mitochondrial function in health and disease. The physiological role of PINK1 in the major cells involved in diabetic kidney disease including proximal tubular cells and podocytes will also be summarized. Collectively, these studies suggested that targeting PINK1 may offer a promising alternative for the treatment of diabetic kidney disease. Keywords: PINK1; diabetic kidney disease; mitochondria; mitochondria quality control; mitophagy Citation: Huang, C.; Bian, J.; Cao, Q.; 1. Introduction Chen, X.-M.; Pollock, C.A. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Systems Analysis Implicates WAVE2&Nbsp
JACC: BASIC TO TRANSLATIONAL SCIENCE VOL.5,NO.4,2020 ª 2020 THE AUTHORS. PUBLISHED BY ELSEVIER ON BEHALF OF THE AMERICAN COLLEGE OF CARDIOLOGY FOUNDATION. THIS IS AN OPEN ACCESS ARTICLE UNDER THE CC BY-NC-ND LICENSE (http://creativecommons.org/licenses/by-nc-nd/4.0/). PRECLINICAL RESEARCH Systems Analysis Implicates WAVE2 Complex in the Pathogenesis of Developmental Left-Sided Obstructive Heart Defects a b b b Jonathan J. Edwards, MD, Andrew D. Rouillard, PHD, Nicolas F. Fernandez, PHD, Zichen Wang, PHD, b c d d Alexander Lachmann, PHD, Sunita S. Shankaran, PHD, Brent W. Bisgrove, PHD, Bradley Demarest, MS, e f g h Nahid Turan, PHD, Deepak Srivastava, MD, Daniel Bernstein, MD, John Deanfield, MD, h i j k Alessandro Giardini, MD, PHD, George Porter, MD, PHD, Richard Kim, MD, Amy E. Roberts, MD, k l m m,n Jane W. Newburger, MD, MPH, Elizabeth Goldmuntz, MD, Martina Brueckner, MD, Richard P. Lifton, MD, PHD, o,p,q r,s t d Christine E. Seidman, MD, Wendy K. Chung, MD, PHD, Martin Tristani-Firouzi, MD, H. Joseph Yost, PHD, b u,v Avi Ma’ayan, PHD, Bruce D. Gelb, MD VISUAL ABSTRACT Edwards, J.J. et al. J Am Coll Cardiol Basic Trans Science. 2020;5(4):376–86. ISSN 2452-302X https://doi.org/10.1016/j.jacbts.2020.01.012 JACC: BASIC TO TRANSLATIONALSCIENCEVOL.5,NO.4,2020 Edwards et al. 377 APRIL 2020:376– 86 WAVE2 Complex in LVOTO HIGHLIGHTS ABBREVIATIONS AND ACRONYMS Combining CHD phenotype–driven gene set enrichment and CRISPR knockdown screening in zebrafish is an effective approach to identifying novel CHD genes. -

Primate Specific Retrotransposons, Svas, in the Evolution of Networks That Alter Brain Function
Title: Primate specific retrotransposons, SVAs, in the evolution of networks that alter brain function. Olga Vasieva1*, Sultan Cetiner1, Abigail Savage2, Gerald G. Schumann3, Vivien J Bubb2, John P Quinn2*, 1 Institute of Integrative Biology, University of Liverpool, Liverpool, L69 7ZB, U.K 2 Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, The University of Liverpool, Liverpool L69 3BX, UK 3 Division of Medical Biotechnology, Paul-Ehrlich-Institut, Langen, D-63225 Germany *. Corresponding author Olga Vasieva: Institute of Integrative Biology, Department of Comparative genomics, University of Liverpool, Liverpool, L69 7ZB, [email protected] ; Tel: (+44) 151 795 4456; FAX:(+44) 151 795 4406 John Quinn: Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, The University of Liverpool, Liverpool L69 3BX, UK, [email protected]; Tel: (+44) 151 794 5498. Key words: SVA, trans-mobilisation, behaviour, brain, evolution, psychiatric disorders 1 Abstract The hominid-specific non-LTR retrotransposon termed SINE–VNTR–Alu (SVA) is the youngest of the transposable elements in the human genome. The propagation of the most ancient SVA type A took place about 13.5 Myrs ago, and the youngest SVA types appeared in the human genome after the chimpanzee divergence. Functional enrichment analysis of genes associated with SVA insertions demonstrated their strong link to multiple ontological categories attributed to brain function and the disorders. SVA types that expanded their presence in the human genome at different stages of hominoid life history were also associated with progressively evolving behavioural features that indicated a potential impact of SVA propagation on a cognitive ability of a modern human. -

PINK1, Parkin, and DJ-1 Mutations in Italian Patients with Early-Onset Parkinsonism
European Journal of Human Genetics (2005) 13, 1086–1093 & 2005 Nature Publishing Group All rights reserved 1018-4813/05 $30.00 www.nature.com/ejhg ARTICLE PINK1, Parkin, and DJ-1 mutations in Italian patients with early-onset parkinsonism Christine Klein*,1,2,9, Ana Djarmati1,2,3,9, Katja Hedrich1,2, Nora Scha¨fer1,2, Cesa Scaglione4, Roberta Marchese5, Norman Kock1,2, Birgitt Schu¨le1,2, Anja Hiller1, Thora Lohnau1,2, Susen Winkler1,2, Karin Wiegers1,2, Robert Hering6, Peter Bauer6, Olaf Riess6, Giovanni Abbruzzese5, Paolo Martinelli4 and Peter P Pramstaller7,8 1Department of Neurology, University of Lu¨beck, Lu¨beck, Germany; 2Department of Human Genetics, University of Lu¨beck, Lu¨beck, Germany; 3Faculty of Biology, University of Belgrade, Belgrade, Serbia; 4Institute of Neurology, University of Bologna, Bologna, Italy; 5Department of Neurology, University of Genova, Genova, Italy; 6Department of Medical Genetics, University of Tu¨bingen, Tu¨bingen, Germany; 7Department of Neurology, General Regional Hospital, Bolzano-Bozen, Italy; 8Department of Genetic Medicine, EURAC-Research, Bolzano-Bozen, Italy Recessively inherited early-onset parkinsonism (EOP) has been associated with mutations in the Parkin, DJ-1, and PINK1 genes. We studied the prevalence of mutations in all three genes in 65 Italian patients (mean age of onset: 43.275.4 years, 62 sporadic, three familial), selected by age at onset equal or younger than 51 years. Clinical features were compatible with idiopathic Parkinson’s disease in all cases. To detect small sequence alterations in Parkin, DJ-1, and PINK1, we performed a conventional mutational analysis (SSCP/ dHPLC/sequencing) of all coding exons of these genes. -

Genetic and Genomic Analysis of Hyperlipidemia, Obesity and Diabetes Using (C57BL/6J × TALLYHO/Jngj) F2 Mice
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Nutrition Publications and Other Works Nutrition 12-19-2010 Genetic and genomic analysis of hyperlipidemia, obesity and diabetes using (C57BL/6J × TALLYHO/JngJ) F2 mice Taryn P. Stewart Marshall University Hyoung Y. Kim University of Tennessee - Knoxville, [email protected] Arnold M. Saxton University of Tennessee - Knoxville, [email protected] Jung H. Kim Marshall University Follow this and additional works at: https://trace.tennessee.edu/utk_nutrpubs Part of the Animal Sciences Commons, and the Nutrition Commons Recommended Citation BMC Genomics 2010, 11:713 doi:10.1186/1471-2164-11-713 This Article is brought to you for free and open access by the Nutrition at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Nutrition Publications and Other Works by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. Stewart et al. BMC Genomics 2010, 11:713 http://www.biomedcentral.com/1471-2164/11/713 RESEARCH ARTICLE Open Access Genetic and genomic analysis of hyperlipidemia, obesity and diabetes using (C57BL/6J × TALLYHO/JngJ) F2 mice Taryn P Stewart1, Hyoung Yon Kim2, Arnold M Saxton3, Jung Han Kim1* Abstract Background: Type 2 diabetes (T2D) is the most common form of diabetes in humans and is closely associated with dyslipidemia and obesity that magnifies the mortality and morbidity related to T2D. The genetic contribution to human T2D and related metabolic disorders is evident, and mostly follows polygenic inheritance. The TALLYHO/ JngJ (TH) mice are a polygenic model for T2D characterized by obesity, hyperinsulinemia, impaired glucose uptake and tolerance, hyperlipidemia, and hyperglycemia. -

2328.Full.Pdf
Altered interplay between endoplasmic reticulum and mitochondria in Charcot–Marie–Tooth type 2A neuropathy Nathalie Bernard-Marissala,b,1, Gerben van Hamerenc, Manisha Junejad,e, Christophe Pellegrinof, Lauri Louhivuorig, Luca Bartesaghih,i, Cylia Rochata, Omar El Mansoura, Jean-Jacques Médardh,i, Marie Croisierj, Catherine Maclachlanj, Olivier Poirotk, Per Uhléng, Vincent Timmermand,e, Nicolas Tricaudc, Bernard L. Schneidera,1,2, and Roman Chrasth,i,1,2 aBrain Mind Institute, École Polytechnique Fédérale de Lausanne, 1015 Lausanne, Switzerland; bMarseille Medical Genetics, INSERM, Aix-Marseille Univ, 13385 Marseille, France; cINSERM U1051, Institut des Neurosciences de Montpellier, Université de Montpellier, 34295 Montpellier, France; dPeripheral Neuropathy Research Group, Department of Biomedical Sciences, University of Antwerp, 2610 Antwerp, Belgium; eInstitute Born Bunge, 2610 Antwerp, Belgium; fInstitut de Neurobiologie de la Méditerranée, INSERM, Aix-Marseille Univ, 13009 Marseille, France; gDepartment of Medical Biochemistry and Biophysics, Karolinska Institutet, 17177 Stockholm, Sweden; hDepartment of Neuroscience, Karolinska Institutet, 17177 Stockholm, Sweden; iDepartment of Clinical Neuroscience, Karolinska Institutet, 17177 Stockholm, Sweden; jCentre of Interdisciplinary Electron Microscopy, École Polytechnique Fédérale de Lausanne, 1015 Lausanne, Switzerland; and kDepartment of Medical Genetics, University of Lausanne, 1005 Lausanne, Switzerland Edited by Stephen T. Warren, Emory University School of Medicine, Atlanta, GA, and approved December 14, 2018 (received for review June 26, 2018) Mutations in the MFN2 gene encoding Mitofusin 2 lead to the axons (8). However, the long-term progression of the disease and development of Charcot–Marie–Tooth type 2A (CMT2A), a domi- the mechanisms underlying motor and/or sensory dysfunction nant axonal form of peripheral neuropathy. Mitofusin 2 is local- have not been fully characterized in this model. -

Mitoq Inhibits Hepatic Stellate Cell Activation and Liver Fibrosis by Enhancing PINK1/Parkin-Mediated Mitophagy
MitoQ Inhibits Hepatic Stellate Cell Activation and Liver Fibrosis by Enhancing PINK1/parkin-mediated Mitophagy Shi-Ying Dou Second Hospital of Hebei Medical University Jiu-Na Zhang Second Hospital of Hebei Medical University Xiao-Li Xie Second Hospital of Hebei Medical University Ting Liu Second Hospital of Hebei Medical University Jun-Li Hu Second Hospital of Hebei Medical University Xiao-Yu Jiang Second Hospital of Hebei Medical University Miao-Miao Wang Second Hospital of Hebei Medical University Hui-Qing Jiang ( [email protected] ) Second Hospital of Hebei Medical University https://orcid.org/0000-0002-5235-1457 Research Article Keywords: Liver brosis, hepatic stellate cell, ubiquinone, PINK1 mitophagy Posted Date: March 30th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-344963/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/18 Abstract Mitophagy plays an important role in the activation of hepatic stellate cells (HSCs). Mitochondria- targeted ubiquinone (MitoQ) is a mitochondria-targeted antioxidant that reduces the production of intracellular reactive oxygen species (ROS). However, its relationship with mitophagy remains unclear. This study evaluated mitophagy during HSC activation and the effects of MitoQ on mitophagy in cell culture and in an animal model of the activation of HSCs. We found that MitoQ reduced the activation of HSCs and alleviated hepatic brosis. While activation of primary HSCs or LX-2 cells was associated with reduced PINK1/parkin-mediated mitophagy, MitoQ reduced intracellular ROS levels, enhanced PINK1/parkin-mediated mitophagy, and inhibited the activation of HSCs. After knocking down the key mitophagy-related protein, PINK1, in LX-2 cells to block mitophagy, MitoQ intervention failed to inhibit HSC activation.