Optimized Synthesis of 3′-O-Aminothymidine and Evaluation of Its Oxime Derivative As an Anti-HIV Agent
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

United States Patent (19) 11 Patent Number: 5,945,382 Cantegrill Et Al
US005.945382A United States Patent (19) 11 Patent Number: 5,945,382 Cantegrill et al. (45) Date of Patent: *Aug. 31, 1999 54 FUNGICIDAL ARYLPYRAZOLES 2300173 12/1990 Japan. 2224208 5/1990 United Kingdom. 75 Inventors: Richard Cantegril, Lyons; Denis Croisat, Paris; Philippe Desbordes, OTHER PUBLICATIONS Lyons, Francois Guigues, English translation of JP 2-300173, 1990. Rillieux-la-Pape; Jacques Mortier, La English translation of JP 59–53468, 1984. Bouéxier; Raymond Peignier, Caluire; English translation of JP 3-93774, 1991. Jean Pierre Vors, Lyons, all of France Miura et al., (CA 1.14:164226), 1991. Miura et al., (CA 115:92260), 1991. 73 Assignee: Rhone-Poulenc Agrochimie, Lyons, Chemical Abstracts, vol. 108, No. 23, 1986, abstract No. France 204577b. CAS Registry Handbook, No. section, RN=114913-44-9, * Notice: This patent is subject to a terminal dis 114486-01-0, 99067-15-9, 113140-19-5, 73227-97-1, claimer. 27069-17-6, 18099-21–3, 17978-27-7, 1988. 21 Appl. No.: 08/325,283 Hattori et al., CA 68:68981 (1968), Registry No. 17978–25–5, 17978-26-6, 17978-27-7 and 18099–21-3. 22 PCT Filed: Apr. 26, 1993 Hattori et al., CA 68:68982 (1968), Registry No. 17978-28-8. 86 PCT No.: PCT/FR93/00403 Janssen et al., CA 78: 159514 (1973), Registry No. S371 Date: Dec. 22, 1994 38858-97-8 and 38859-02-8. Chang et al., CA 92:146667 (1980), Registry No. S 102(e) Date: Dec. 22, 1994 73227 91-1. Berenyi et al., CA 94:156963 (1981), Registry No. -

The Evolution of Pleconaril: Modified O-Alkyl Linker Analogs Have
molecules Communication The Evolution of Pleconaril: Modified O-Alkyl Linker Analogs Have Biological Activity towards Coxsackievirus B3 Nancy 1, 2, 1 3 Alexandrina Volobueva y, Anna Egorova y, Anastasia Galochkina , Sean Ekins , Vladimir Zarubaev 1 and Vadim Makarov 2,* 1 Saint-Petersburg Pasteur Institute, Mira str., 14, 197101 Saint Petersburg, Russia; [email protected] (A.V.); [email protected] (A.G.); [email protected] (V.Z.) 2 Bach Institute of Biochemistry, Research Center of Biotechnology of the Russian Academy of Sciences, Leninsky prospect, 33, build. 2, 119071 Moscow, Russia; [email protected] 3 Collaborations Pharmaceuticals, Inc., 840 Main Campus Drive, Lab 3510, Raleigh, NC 27606, USA; [email protected] * Correspondence: [email protected] These authors contributed equally to this work. y Received: 10 February 2020; Accepted: 13 March 2020; Published: 16 March 2020 Abstract: Coxsackieviruses type B are one of the most common causes of mild upper respiratory and gastrointestinal illnesses. At the time of writing, there are no approved drugs for effective antiviral treatment for Coxsackieviruses type B. We used the core-structure of pleconaril, a well-known antienteroviral drug candidate, for the synthesis of novel compounds with O-propyl linker modifications. Some original compounds with 4 different linker patterns, such as sulfur atom, ester, amide, and piperazine, were synthesized according to five synthetic schemes. The cytotoxicity and bioactivity of 14 target compounds towards Coxsackievirus B3 Nancy were examined. Based on the results, the values of 50% cytotoxic dose (CC50), 50% virus-inhibiting dose (IC50), and selectivity index (SI) were calculated for each compound. Several of the novel synthesized derivatives exhibited a strong anti-CVB3 activity (SI > 20 to > 200). -

Anhydrous Hydration of Nitriles to Amides: P-Carbomethoxybenzamide
A Publication of Reliable Methods for the Preparation of Organic Compounds Working with Hazardous Chemicals The procedures in Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices. In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red “Caution Notes” within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices. The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein. -

New York City Department of Environmental Protection Community Right-To-Know: List of Hazardous Substances
New York City Department of Environmental Protection Community Right-to-Know: List of Hazardous Substances Updated: 12/2015 Definitions SARA = The federal Superfund Amendments and Reauthorization Act (enacted in 1986). Title III of SARA, known as the Emergency Planning and Community Right-to-Act, sets requirements for hazardous chemicals, improves the public’s access to information on chemical hazards in their community, and establishes reporting responsibilities for facilities that store, use, and/or release hazardous chemicals. RQ = Reportable Quantity. An amount entered in this column indicates the substance may be reportable under §304 of SARA Title III. Amount is in pounds, a "K" represents 1,000 pounds. An asterisk following the Reporting Quantity (i.e. 5000*) will indicate that reporting of releases is not required if the diameter of the pieces of the solid metal released is equal to or exceeds 100 micrometers (0.004 inches). TPQ = Threshold Planning Quantity. An amount entered in this column reads in pounds and indicates the substance is an Extremely Hazardous Substance (EHS), and may require reporting under sections 302, 304 & 312 of SARA Title III. A TPQ with a slash (/) indicates a "split" TPQ. The number to the left of the slash is the substance's TPQ only if the substance is present in the form of a fine powder (particle size less than 100 microns), molten or in solution, or reacts with water (NFPA rating = 2, 3 or 4). The TPQ is 10,000 lb if the substance is present in other forms. A star (*) in the 313 column= The substance is reportable under §313 of SARA Title III. -

Efforts Towards the Synthesis of Stelliferin Natural Products Michelle
Efforts towards the Synthesis of Stelliferin Natural Products A thesis presented by Michelle H. Fisher A.B., Chemistry (1994) Princeton University to The Department of Chemistry in partial fulfillment of the requirements for the degree of Masters of Science at the Massachusetts Institute of Technology June 1997 © 1997 Massachusetts Institute of Technology. All Rights Reserved. The author hereby grants MIT permission to reproduce and to distribute publicly paper and electronic copies of this thesis document in whole or in part. Signature of Author: Department ot Chemistry May 2, 1997 Certified by: Scott Vkgil Thesis Advisor Accepted by Dietmar Seyferth Chairman, Departmental Committee on Graduate Students 9 ?W Rg ~~ i L::::~~ Efforts towards the Synthesis of Stelliferin Natural Products by Michelle H. Fisher Submitted to the Department of Chemistry on May 2, 1997 in partial fulfillment of the requirements for the degree of Masters of Science in Chemistry at the Massachusetts Institute of Technology. ABSTRACT Studies for this thesis involve efforts toward the synthesis of stelliferin natural product Stellettin A (2). Key steps towards this synthesis were developed using epiandrosterone (9) as a model for the stelliferin ring system. Our synthetic strategy produced new functionalized steroid compounds, including 15, as well as more efficient syntheses of various 2-pyrone derivatives. The metal-catalyzed coupling of 16 and 5 resulted in a novel steroidal compound 27. Hydrogenation of this molecule afforded the Stellettin A model 10. The methodology for synthesis of Stellettin A (2) thus has been developed, which can be utilized on the actual stelliferin ring system. 0 O 0 f ..HO"' v 0 Thesis Supervisor: Scott C. -

The Dipole Moments and Molecular Structure of Formyl and Acetyl Compounds
THE DIPOLE MOMENTS AND MOLECULAR STRUCTURE OF FORMYL AND ACETYL COMPOUNDS BY R. RAK~,N AND SUNDARF_8A SOUNDARARAJAN (Department of General Chemistry, Indian lnstitute of Scienr Banstalore-3) Received March 28, 1958 (Communicated by Prof. K. R. Krishnaswami, F.A.SC.) INTRODUCTION DtmrNG the past two decades the concept of hyperconjugation has been employed to interpret and to correlate a variety of experimental facts. ~' s Since this was explicitly achieved by Baker and Nathan 1 to explain the ano- malous behaviour of certain series of alkyl substituted compounds, this pheno- menon has been subjected to a quantitative study by the method of molecular orbitals ~o and also by means of study of its effects on properties like bond length, heats of hydrogenation and dipole moment. Dipole moment studies have given charaiteristic evidence of hyperconjugation in aldehydes and nitfiles a chloroform and methyl chloroform, x7 acrylonitrile and isocrotyl chlorideTM and some unsaturated aldehydes, ethers, and halogen compounds, ts Since this problem has not been adequately considered from the point of view of dipole moments a study of the eleet¡ moments of a series of com- pounds has been earried out with a view to investigate the extent of hyper- conjugation in these compounds. Particular compounds for which dieleetrie measurements have been made are the following: Formhydrazide, Aeethydra- zide, Acetaldoxime, Acetoxime and Formamidoxime. EXPERIMENTAL Materials Dioxan.--Dioxan which was the solvent employed in this work was purified by Eigenberger's methodxl the details of which are given in an earlier paper. ~a d~~.c. ----- 1.01690, %ss.c. = 2.1890. Forro and Acethydrazides.--The two hydrazides are prepared according to the method of Sch5fer and Schwan. -

Table of Contents
MONTANA TECH i CHEMICAL HYGIENE PLAN TABLE OF CONTENTS TABLE OF CONTENTS .......................................................................................... i STATEMENT OF POLICY .................................................................................... 1 CHEMICAL HYGIENE RESPONSIBILITIES ........................................................ 2 LABORATORY FACILITIES ................................................................................. 4 Maintenance ..................................................................................................... 4 Evaluation ......................................................................................................... 5 CONTROL MEASURES ....................................................................................... 5 Work Habits ...................................................................................................... 5 Laboratory Hygiene ........................................................................................... 6 Engineering Controls ........................................................................................ 6 Instrument Equipment and Use ........................................................................ 7 Personal Protective Equipment (PPE) .............................................................. 8 Eye Protection ............................................................................................. 8 Hand Protection .......................................................................................... -

L-G-0009995667-0020105508.Pdf
Handbook of Reagents for Organic Synthesis Reagents for Heteroarene Synthesis OTHER TITLES IN THIS COLLECTION Reagents for Organocatalysis Edited by Tomislav Rovis ISBN 978 1 119 06100 7 Reagents for Heteroarene Functionalization Edited by Andre´ Charette ISBN 978 1 118 72659 4 Catalytic Oxidation Reagents Edited by Philip L. Fuchs ISBN 978 1 119 95327 2 Reagents for Silicon-Mediated Organic Synthesis Edited by Philip L. Fuchs ISBN 978 0 470 71023 4 Sulfur-Containing Reagents Edited by Leo A. Paquette ISBN 978 0 470 74872 5 Reagents for Radical and Radical Ion Chemistry Edited by David Crich ISBN 978 0 470 06536 5 Catalyst Components for Coupling Reactions Edited by Gary A. Molander ISBN 978 0 470 51811 3 Fluorine-Containing Reagents Edited by Leo A. Paquette ISBN 978 0 470 02177 4 Reagents for Direct Functionalization for C–H Bonds Edited by Philip L. Fuchs ISBN 0 470 01022 3 Reagents for Glycoside, Nucleotide, and Peptide Synthesis Edited by David Crich ISBN 0 470 02304 X Reagents for High-Throughput Solid-Phase and Solution-Phase Organic Synthesis Edited by Peter Wipf ISBN 0 470 86298 X Chiral Reagents for Asymmetric Synthesis Edited by Leo A. Paquette ISBN 0 470 85625 4 Activating Agents and Protecting Groups Edited by Anthony J. Pearson and William R. Roush ISBN 0 471 97927 9 Acidic and Basic Reagents Edited by Hans J. Reich and James H. Rigby ISBN 0 471 97925 2 Oxidizing and Reducing Agents Edited by Steven D. Burke and Rick L. Danheiser ISBN 0 471 97926 0 Reagents, Auxiliaries, and Catalysts for C–C Bond Formation Edited by Robert M. -

Related Properties from Molecular Structures
Electronic Supplementary Material (ESI) for Green Chemistry. This journal is © The Royal Society of Chemistry 2021 1 SUPPORTING INFORMATION 2 A multi-task deep learning neural network for predicting flammability- 3 related properties from molecular structures 4 Ao Yang‡a,b, Yang Su‡c,a, Zihao Wanga, Saimeng Jina, Jingzheng Renb, Xiangping Zhangd, 5 Weifeng Shen*a and James H. Clarke 6 aSchool of Chemistry and Chemical Engineering, Chongqing University, Chongqing 400044, China 7 bDepartment of Industrial and Systems Engineering, The Hong Kong Polytechnic University, Hong 8 Kong SAR, China 9 cSchool of Intelligent Technology and Engineering, Chongqing University of Science and Technology, 10 Chongqing 401331, China 11 dBeijing Key Laboratory of Ionic Liquids Clean Process, CAS Key Laboratory of Green Process and 12 Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing, 100190, China 13 eGreen Chemistry Centre of Excellence, University of York, York YO105D, UK 14 *Correspondence should be addressed to: Weifeng Shen ([email protected]) 15 ‡These authors contributed equally to this work. 16 S1. Signature descriptors 17 The signature descriptor1 is an effective tool for encoding molecular structures with all 18 atomic neighborhood into strings. This descriptor involving two forms: the first one is the 19 atomic signatures generated at a specified root atom in a molecule, the other one is the molecular 20 signatures combined from all atom signatures linearly. The size of each atomic signature is 21 determined by a parameter named “height”, h. An atomic Signature is a tree subgraph including 22 all atoms/bonds extending out to the predefined distance h without backtracking. -

United States Patent Office Patented Apr
3,658,869 United States Patent Office Patented Apr. 25, 1972 1. 2 Separation difficulties of organic solvent media since the 3,658,869 1-hydrocarbylthio-aldoximes precipitate out of the aque PROCESS FOR PREPARING SULFUR CONTAINING ALDOX MES ous medium, being only slightly soluble in water. Further Samuel B. Soloway, Modesto, Calif., and Herbert P. more, the use of water as the reaction medium obviates Rosinger, Kent, England, assignors to Shell Oil Com any need for changing solvent systems or for isolating the pany, New York, N.Y. intermediate halogenation product. This reduction in op No Drawing. Continuation-in-part of application Ser. No. erating steps leads to the higher yields necessary for 645,511, June 12, 1967. This application Aug. 25, 1969, commercial operations. The improved process of the Ser. No. 852,899 invention has consistently produced yields of at least 80% Claims priority, application Great Britain, June 13, 1966, O While a similar process using organic solvents seldom pro 26,258/66 duced yields of about 50%. The costs are also greatly Int, C. C07 131/00 reduced not only because of the use of water but also in U.S. C. 260-453 R 9 Claims the elimination of added costs connected with handling toxic, volatile organic solvents. Since the chlorination of the aldoxime is carried out in ABSTRACT OF THE DISCLOSURE an aqueous medium it is essential that the aldoxime re Process for preparing 1-hydrocarbythio-aldoximes in an actant be at least substantially soluble in water. The crude aqueous reaction medium by the halogenation of an al reaction mixture from the chlorination then is reacted doxime followed by reaction with a mercaptain in the with the mercaptan. -

DIPPR Compound List-2011
DIPPR Compound List 2011 Sponsor ChemID Chemical Name Formula CAS# Only 1 METHANE CH4 74‐82‐8 2 ETHANE C2H6 74‐84‐0 3 PROPANE C3H8 74‐98‐6 4 ISOBUTANE C4H10 75‐28‐5 5n‐BUTANE C4H10 106‐97‐8 6 1,1,3‐TRIMETHYLCYCLOHEXANE C9H18 3073‐66‐3S 7n‐PENTANE C5H12 109‐66‐0 8 ISOPENTANE C5H12 78‐78‐4 9 NEOPENTANE C5H12 463‐82‐1 11 n‐HEXANE C6H14 110‐54‐3 12 2‐METHYLPENTANE C6H14 107‐83‐5 13 3‐METHYLPENTANE C6H14 96‐14‐0 14 2,2‐DIMETHYLBUTANE C6H14 75‐83‐2 15 2,3‐DIMETHYLBUTANE C6H14 79‐29‐8 17 n‐HEPTANE C7H16 142‐82‐5 18 2‐METHYLHEXANE C7H16 591‐76‐4 19 3‐METHYLHEXANE C7H16 589‐34‐4 20 3‐ETHYLPENTANE C7H16 617‐78‐7 21 2,2‐DIMETHYLPENTANE C7H16 590‐35‐2 22 2,3‐DIMETHYLPENTANE C7H16 565‐59‐3 23 2,4‐DIMETHYLPENTANE C7H16 108‐08‐7 24 3,3‐DIMETHYLPENTANE C7H16 562‐49‐2 25 2,2,3‐TRIMETHYLBUTANE C7H16 464‐06‐2 27 n‐OCTANE C8H18 111‐65‐9 28 2‐METHYLHEPTANE C8H18 592‐27‐8 29 3‐METHYLHEPTANE C8H18 589‐81‐1 30 4‐METHYLHEPTANE C8H18 589‐53‐7 31 3‐ETHYLHEXANE C8H18 619‐99‐8 32 2,2‐DIMETHYLHEXANE C8H18 590‐73‐8 33 2,3‐DIMETHYLHEXANE C8H18 584‐94‐1 34 2,4‐DIMETHYLHEXANE C8H18 589‐43‐5 35 2,5‐DIMETHYLHEXANE C8H18 592‐13‐2 36 3,3‐DIMETHYLHEXANE C8H18 563‐16‐6 37 3,4‐DIMETHYLHEXANE C8H18 583‐48‐2 38 2‐METHYL‐3‐ETHYLPENTANE C8H18 609‐26‐7 39 3‐METHYL‐3‐ETHYLPENTANE C8H18 1067‐08‐9 40 2,2,3‐TRIMETHYLPENTANE C8H18 564‐02‐3 41 2,2,4‐TRIMETHYLPENTANE C8H18 540‐84‐1 42 2,3,3‐TRIMETHYLPENTANE C8H18 560‐21‐4 43 2,3,4‐TRIMETHYLPENTANE C8H18 565‐75‐3 44 2,2,3,3‐TETRAMETHYLBUTANE C8H18 594‐82‐1 46 n‐NONANE C9H20 111‐84‐2 47 2,2,5‐TRIMETHYLHEXANE C9H20 3522‐94‐9 48 3,3,5‐TRIMETHYLHEPTANE -
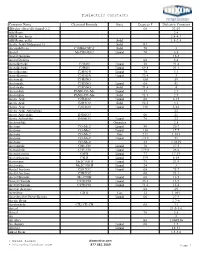
Dielectric Constants
Dielectric Constants Common Name Chemical Formula State Degrees F Dielectric Constant (Ethylenedioxy)diethanol-2,2` 68 23.69 ABS Resin 2.4 ABS Resin, lump 2.4-4.1 ABS Resin, pellet Solid 1.5-2.5 Acatic.Acid (36degrees F) Solid 4.1 Acenaphthene C10H6(CH2)2 Solid 70 3 Acetal MeCH(OEt)2 Liquid 70 3.6 Acetal Bromide 16.5 Acetal Doxime 68 3.4 Acetaldehyde C2H4O Liquid 50 21.8 Acetaldehyde C2H4O Liquid 69.8 21.1 Acetaldoxime C2H5ON Liquid 70 3.4 Acetaldoxime C2H5ON Liquid 73.4 3 Acetamide C3H5NO 180 59 Acetamide C3H5NO Liquid 68 41 Acetamide C3H5NO Solid 71.6 4 Acetanilide PhNH-CO-Me Liquid 71 2.9 Acetanilide PhNH-CO-Me Solid 71.6 2.9 Acetic Acid C2H4O2 Liquid 68 6.15 Acetic Acid C2H4O2 Solid 32.5 4.1 Acetic Acid C2H4O2 Liquid 158 6.62 Acetic Acid, Anhydrous 22 Acetic Anhydride H4H6O3 66 21 Acetic Anhydride H4H6O3 Liquid 70 22 Acetoanilide Granules 2.8 Acetone CO-Me2 Liquid 80 20.7 Acetone CO-Me2 Liquid 130 17.7 Acetone CO-Me2 Gas 212 1.015 Acetone CO-Me2 Liquid -112 31 Acetone CO-Me2 32 1.0159 Acetonitrile CH3-CN Liquid 70 37.5 Acetonitrile CH3-CN Liquid 179.6 26.6 Acetophenone C8H8 Liquid 77 17.39 Acetophenone C8H8 Liquid 394 8.64 Acetoxime Me2C:NOH Liquid 75 23.9 Acetoxime Me2C:NOH Liquid 24 3 Acetyl Acetone C5H2O2 Liquid 68 25.7 Acetyl Acetone C5H2O2 68 23.1 Acetyl Bromide Me-COBr Liquid 68 16.5 Acetyl Chloride C2H3ClO Liquid 35.6 16.9 Acetyl Chloride C2H3ClO Liquid 71.6 15.8 Acetyle Acetone 68 25 Acetylene C2H2 Gas 32 1.001 Acetylmethyl Hexyl Ketone Liquid 66 27.9 Acrylic Resin 2.7-6 Acrylonitrile CH2:CH-CN 68 33 Acteal 21.0-3.6 Acteal