The Importance of Iminium Geometry Control in Enamine Catalysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Alkylation by Enamines for Synthesis of Some Heterocyclic Compounds ﻴﺩ
------J. Raf. Sci., Vol. 20, No.2, pp 92- 101, 2009 ------ Alkylation by Enamines for Synthesis of some Heterocyclic Compounds Jasim A. Abdullah Department of Chemistry College of Education Mosul University (Received 25/ 9/ 2008 ; Accepted 13 / 4 / 2009) ABSTRACT Compounds of 4-phenyl-3-butene-2-one (1) and 4-(4-chlorophenyl)-3-butene-2-one (2) were prepared by reaction of benzaldehyde or 4-chlorobenzaldehyde with acetone. Also 1,3-diphenyl-2-chloropropene-1-one (3) was synthesized from the reaction of benzaldehyde with 2-chloroacetophenone through Claisen-Shmidt condensation. The substituted ∆1(9)- octalone-2 (4,5) was prepared by reaction of the compounds (1and2) with cyclohexanone through Michael addition followed by aldol condensation. Compounds (4,5) were reacted with alkyl halide, as 2-chloroacetophenone or 1,3-diphenyl-2-chloropropene-1-one (3), via enamines formation which then hydrolyzed to give compounds (6-9). Compounds (6,7) were reacted with hydrazine , urea and thiourea to afford compounds (10-15) . The structures of all synthesized compounds were confirmed by available physical and spectral means . Keywords : Enamines , Heterocyclic compounds. ــــــــــــــــــــــــــــــــــــــــــــــــــ ﺍﻻﻟﻜﻠﺔ ﺒﻭﺍﺴﻁﺔ ﺍﻷﻴﻨﺎﻤﻴﻨﺎﺕ ﻟﺘﺸﻴﻴﺩ ﺒﻌﺽ ﺍﻟﻤﺭﻜﺒﺎﺕ ﺍﻟﺤﻠﻘﻴﺔ ﻏﻴﺭ ﺍﻟﻤﺘﺠﺎﻨﺴﺔ ﺍﻟﻤﻠﺨﺹ ﺘﻡ ﺘﺤﻀﻴﺭ ﺍﻟﻤﺭﻜﺒﺎﺕ 4-ﻓﻨﻴل-3-ﺒﻴﻭﺘﻴﻥ-2-ﺍﻭﻥ (1) ﻭ4-(4-ﻜﻠﻭﺭﻭﻓﻨﻴل)-3-ﺒﻴﻭﺘﻴﻥ-2-ﺍﻭﻥ (2) ﻤﻥ ﺘﻔﺎﻋل ﺍﻟﺒﻨﺯﺍﻟﺩﻴﻬﻴﺩ ﺃﻭ 4-ﻜﻠﻭﺭﻭﺍﻟﺒﻨﺯﺍﻟﺩﻴﻬﻴﺩ ﻤﻊ ﺍﻻﺴﻴﺘﻭﻥ . ﺤﻀﺭ ﺍﻟﻤﺭﻜﺏ 3,1-ﺜﻨﺎﺌﻲ ﻓﻨﻴل -2-ﻜﻠـﻭﺭﻭﺒﺭﻭﺒﻴﻥ -1-ﺍﻭﻥ (3) ﻤـﻥ ﺘﻔﺎﻋـل ﺍﻟﺒﻨﺯﺍﻟﺩﻴﻬﻴـﺩ ﻤـﻊ -2 9 1 ﻜﻠﻭﺭﻭﺍﺴﻴﺘﻭﻓﻴﻨﻭﻥ ﺒﻭﺴﺎﻁﺔ ﺘﻜﺎﺜﻑ (ﻜﻠﻴﺯﻥ-ﺸﻤﺩﺕ).ﺤﻀﺭﺕ ﻤﻌﻭﻀﺎﺕ ∆ ( ) –ﺍﻭﻜﺘﺎﻟﻭﻥ-2 ﻤﻥ ﺨﻼل ﺘﻔﺎﻋل ﺍﻟﻤﺭﻜﺒﻴﻥ (2,1) ﻤﻊ ﺍﻟﻬﻜﺴﺎﻨﻭﻥ ﺍﻟﺤﻠﻘﻲ ﺒﻭﺴﺎﻁﺔ ﺍﻀﺎﻓﺔ ﻤﺎﻴﻜل ﺍﻟﺘﻲ ﻴﺘﺒﻌﻬﺎ ﺘﻜﺎﺜﻑ ﺍﻻﻟﺩﻭل . -

Catalytic Direct Asymmetric Michael Reactions
ORGANIC LETTERS 2001 Catalytic Direct Asymmetric Michael Vol. 3, No. 23 Reactions: Taming Naked Aldehyde 3737-3740 Donors Juan M. Betancort and Carlos F. Barbas III* The Skaggs Institute for Chemical Biology and the Department of Molecular Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037 [email protected] Received September 5, 2001 ABSTRACT Direct catalytic enantio- and diastereoselective Michael addition reactions of unmodified aldehydes to nitro olefins using (S)-2-(morpholinomethyl)- pyrrolidine as a catalyst are described. The reactions proceed in good yield (up to 96%) in a highly syn-selective manner (up to 98:2) with enantioselectivities approaching 80%. The resulting γ-formyl nitro compounds are readily converted to chiral, nonracemic 3,4-disubstituted pyrrolidines. The Michael reaction is generally regarded as one of the Typically, carbon nucleophiles that contain an active most efficient carbon-carbon bond forming reactions, and methylene center such as malonic acid esters, â-keto esters, studies concerning this reaction have played an important nitroalkanes, etc. have been studied in the Michael reaction. role in the development of modern synthetic organic Carbonyl compounds, and ketones in particular, have gener- chemistry.1 As the demand for optically active compounds ally only been used as donors following their preactivation has soared in recent years, much progress has been made in by conversion into a more reactive species such as enol or the development of asymmetric variants of this reaction, enamine equivalents.5,6 In these cases, additional synthetic providing for the preparation of Michael adducts with high enantiomeric purity.2 Though remarkable advances have been (3) (a) Chataigner, I.; Gennari, C.; Ongeri, S.; Piarulli, U.; Ceccarelli, S. -

S41467-018-07534-X.Pdf
ARTICLE DOI: 10.1038/s41467-018-07534-x OPEN Differentiation between enamines and tautomerizable imines in the oxidation reaction with TEMPO Xiaoming Jie1, Yaping Shang1, Zhe-Ning Chen 1, Xiaofeng Zhang1, Wei Zhuang1 & Weiping Su1 Enamine and imine represent two of the most common reaction intermediates in syntheses, and the imine intermediates containing α-hydrogen often exhibit the similar reactivity to 1234567890():,; enamines due to their rapid tautomerization to enamine tautomers. Herein, we report that the minor structural difference between the enamine and the enamine tautomer derived from imine tautomerization results in the different chemo- and regioselectivity in the reaction of cyclohexanones, amines and TEMPO: the reaction of primary amines furnishes the formal oxygen 1,2-migration product, α-amino-enones, while the reaction of secondary amines under similar conditions generates exclusively arylamines via consecutive dehydrogenation on the cyclohexyl rings. The 18O-labeling experiment for α-amino-enone formation revealed that TEMPO served as oxygen transfer reagent. Experimental and computational studies of reaction mechanisms revealed that the difference in chemo- and regioselectivity could be ascribed to the flexible imine-enamine tautomerization of the imine intermediate con- taining an α-hydrogen. 1 State Key Laboratory of Structural Chemistry, Center for Excellence in Molecular Synthesis, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, 155 Yangqiao Road West, Fuzhou 350002, China. -

Enamine-Based Organocatalysis with Proline And
Acc. Chem. Res. 2004, 37, 580-591 carbon bond in a stereospecific fashion. These same Enamine-Based Organocatalysis features are typically absent in traditional synthetic asym- with Proline and Diamines: The metric aldol methodologies.2,3 Our ideal synthetic catalyst would be one that could Development of Direct Catalytic generate enamines from any number of aldehydes or ketones and then direct bond-forming reactions with a Asymmetric Aldol, Mannich, wide variety of electrophiles beyond the carbonyl elec- Michael, and Diels-Alder trophiles of the aldol reaction. Further, since these cata- lysts also generate imines as part of their reaction mech- Reactions anism, electrophilic catalysis might facilitate diverse WOLFGANG NOTZ, FUJIE TANAKA, AND reactions with nucleophiles. Such an antibody might be termed an ªopen-active siteº catalytic antibody. Indeed, CARLOS F. BARBAS, III* The Skaggs Institute for Chemical Biology and the we were able to demonstrate that our aldolase antibodies Departments of Chemistry and Molecular Biology, The could catalyze not only a wide range of intra- and Scripps Research Institute, 10550 North Torrey Pines Road, intermolecular aldol reactions but also decarboxylation La Jolla, California 92037 reactions via imine catalysis and certain Michael reactions Received February 2, 2004 as well.2 Significantly for what would become our studies in organocatalysis, in our search for ªopen-active siteº ABSTRACT antibody catalysis, we studied the potential of aldolase Enamines and imines have long been recognized as key intermedi- antibodies to catalyze the addition of ketones to nitrosty- ates in enzyme catalysis, particularly within a class of enzymes renes in Michael reactions, the addition of ketones to organic chemists would very much like to emulate, the aldolases. -
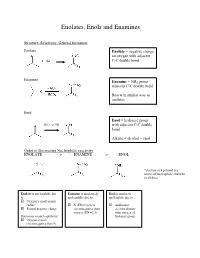
Enolates, Enols and Enamines
Enolates, Enols and Enamines Structure, Reactivity, General formation Enolate Enolate = negative charge on oxygen with adjacent C-C double bond Enamine Enamine = NR2 group adjacent C-C double bond Reacts in similar way as enolates Enol Enol = hydroxyl group - H3O+ or OH with adjacent C-C double bond Alkene + alcohol = enol Order of Decreasing Nucleophilic reactivity ENOLATE > ENAMINE > ENOL *electron-rich pi bond is a source of nucleophilic character in all three Enolate is nucleophilic due Enamine is moderately Enol is moderate to… nucleophilic due to… nucleophile due to… Oxygen’s small atomic radius N (EN=3) is less Additional Formal negative charge electronegative than electron density oxygen (EN =3.5) from oxygen of Detractors to nucleophilicity: hydroxyl group Oxygen is more electronegative than N When looking at which side is favored (products or reactants): Equilibrium favors side with WEAKER acid/base pair Weakest acid and weakest base should be on SAME side When comparing pKa values, juxtapose just the acids or just the bases Side with larger pka value (weaker acid) favored in equilibrium Remember that differences in pKa values reflect a difference quantity of 10x Example: compound H-A (pKa=3) + base compound A (pKa=10) + H-base 7 equilibrium lies to the RIGHT by factor of 10 Some Useful pKa values (from more acidic basic) H2SO4 (pka = -9) H2O (pka = 15.7) + H3O (pka = -1.8) CH3COCH3 (pka = 19) B-diketone (pka = 9) CH3COOCH3 (pKa = 25) HCN (pka = 9.1) LDA-H (pka = 36) CH3OH (pka = 15.5) How do we know which proton to deprotonate? Deprotonate most acidic proton usually leads to the most stable enolate 1. -

New Mechanistic Studies on the Proline-Catalyzed Aldol Reaction
New mechanistic studies on the proline-catalyzed SPECIAL FEATURE aldol reaction Benjamin List*, Linh Hoang, and Harry J. Martin Max-Planck-Institut fu¨r Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mu¨lheim an der Ruhr, Germany Edited by Barry M. Trost, Stanford University, Stanford, CA, and approved February 4, 2004 (received for review December 4, 2003) The mechanism of the proline-catalyzed aldol reaction has stimu- lated considerable debate, and despite limited experimental data, at least five different mechanisms have been proposed. Comple- mentary to recent theoretical studies we have initiated an exper- imental program with the goal of clarifying some of the basic mechanistic questions concerning the proline-catalyzed aldol re- action. Here we summarize our discoveries in this area and provide further evidence for the involvement of enamine intermediates. iscovered in the early 1970s, the Hajos–Parrish–Eder– DSauer–Wiechert reaction (1, 2), a proline-catalyzed in- tramolecular aldol reaction, represents not only the first asym- metric aldol reaction invented by chemists but also the first highly enantioselective organocatalytic transformation [1(4) 3 2(5) 3 3(6)] (Eq. 1 of Scheme 1) (3–6). Inspired by Nature’s phenomenal enzymes, which catalyze direct asymmetric aldol- CHEMISTRY izations of unmodified carbonyl compounds (7, 8), we have recently extended the Hajos–Parrish–Eder–Sauer–Wiechert re- action to the first intermolecular variant (7 ϩ 8 3 9) (Eq. 2 of Scheme 1) (9), and to several other reactions including proline- catalyzed asymmetric Mannich (10), Michael (11), ␣-amination Scheme 1. Proline-catalyzed aldolizations (Eqs. 1–3) and proposed mecha- (12), and intramolecular enolexo aldolization reactions (10 3 nisms (transition states). -

Induction of Optical Activity Via Enamines
University of New Hampshire University of New Hampshire Scholars' Repository Doctoral Dissertations Student Scholarship Spring 1967 INDUCTION OF OPTICAL ACTIVITY VIA ENAMINES PETER WILLIAM KLEINHOMER III. Follow this and additional works at: https://scholars.unh.edu/dissertation Recommended Citation KLEINHOMER, PETER WILLIAM III., "INDUCTION OF OPTICAL ACTIVITY VIA ENAMINES" (1967). Doctoral Dissertations. 854. https://scholars.unh.edu/dissertation/854 This Dissertation is brought to you for free and open access by the Student Scholarship at University of New Hampshire Scholars' Repository. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of University of New Hampshire Scholars' Repository. For more information, please contact [email protected]. This dissertation has been microfilmed exactly as received 67-15,797 KLEINHOMER m , Peter William, 1939- INDUCTION OF OPTICAL ACTIVITY VIA ENAMINES. University of New Hampshire, Ph„D., 1967 Chemistry, organic University Microfilms, Inc., Ann Arbor, Michigan Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. INDUCTION OF OPTICAL ACTIVITY VIA ENAMINES BY PETER WILLIAM KLEINHOMER III B. S., Bloomfield College, I960 M. S., The University of New Hampshire, 1964 A THESIS Submitted to the University of New Hampshire In Partial Fulfillment of The Requirements for the Degree of Doctor of Philosphy Graduate School Department of Chemistry May, 1967 Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. This thesis has been examined and approved. - l i 19 q \ Date Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. ACKNOWLEDGEMENTS The author wishes to thank Dr. Robert E. Lyle, Jr. for his suggestion of the research topic, his consultation and advice. -
1 Preparation and Some Properties
Cambridge University Press 978-0-521-09731-4 - The Chemistry of Enamines S. F. Dyke Excerpt More information 1 Preparation and some properties 1.1. Preparation 1.1.1. From aldehydes and ketones. The most versatile, and most often used method of formation of enamines involves the condensation be- tween an aldehyde or ketone and a secondary amine: H2O Primary amines, of course, also react with carbonyl compounds to form imines: H H C—C=O + H2N— H2O / and whereas tautomerism is possible with the formation of the enamine: the equilibrium is almost always in favour of the imine structure. This book will be concerned almost entirely with tertiary enamines, derived from secondary amines. The commonest procedure for enamine formation, which was first used by Herr and Heyl in 1952, then exploited by Stork et ah, involved heating under reflux an equimolecular mixture of the carbonyl com- pound and the amine in a solvent such as anhydrous benzene, toluene or xylene, with azeotropic removal of the water. Usually a Dean and Stark trap is used, but a number of modifications have been made (e.g. other drying agents, molecular sieves, etc.). Slow reactions can be catalysed by acids, usually ^-toluenesulphonic acid (/?-TSA). However, 1 © in this web service Cambridge University Press www.cambridge.org Cambridge University Press 978-0-521-09731-4 - The Chemistry of Enamines S. F. Dyke Excerpt More information 2 Preparation and some properties in the original preparation of enamines, described in 1936 by Mannich and Davidson, anhydrous potassium carbonate was used as condensing agent. The mechanism of the reaction is usually expressed as shown in scheme 1.1. -
Three-Component Radical Homo Mannich Reaction ✉ Shuai Shi1, Wenting Qiu1, Pannan Miao1, Ruining Li1, Xianfeng Lin1 & Zhankui Sun 1
ARTICLE https://doi.org/10.1038/s41467-021-21303-3 OPEN Three-component radical homo Mannich reaction ✉ Shuai Shi1, Wenting Qiu1, Pannan Miao1, Ruining Li1, Xianfeng Lin1 & Zhankui Sun 1 Aliphatic amine, especially tertiary aliphatic amine, is one of the most popular functionalities found in pharmaceutical agents. The Mannich reaction is a classical and widely used transformation for the synthesis of β-amino-carbonyl products. Due to an ionic nature of the mechanism, the Mannich reaction can only use non-enolizable aldehydes as substrates, which significantly limits the further applications of this powerful approach. Here we show, by 1234567890():,; employing a radical process, we are able to utilize enolizable aldehydes as substrates and develop the three-component radical homo Mannich reaction for the streamlined synthesis of γ-amino-carbonyl compounds. The electrophilic radicals are generated from thiols via the desulfurization process facilitated by visible-light, and then add to the electron-rich double bonds of the in-situ formed enamines to provide the products in a single step. The broad scope, mild conditions, high functional group tolerance, and modularity of this metal-free approach for the synthesis of complex tertiary amine scaffolds will likely be of great utility to chemists in both academia and industry. 1 Shanghai Key Laboratory for Molecular Engineering of Chiral Drugs, School of Pharmacy, Shanghai Jiao Tong University, No. 800 Dongchuan Rd., 200240 ✉ Shanghai, China. email: [email protected] NATURE COMMUNICATIONS | (2021) 12:1006 | https://doi.org/10.1038/s41467-021-21303-3 | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-021-21303-3 mines are very important functional groups in medicinal react with the enolizable carbonyl compound (Fig. -

The Enamine Alkylation and Acylation of Carbonyl Compounds
Jan. N, 1963 ENAMINEALKYLATION AND ACYLATIONOF CARBONYLS 207 Sec. 2. Removal of Protecting Groups.-The conditions for treated with ninhydrin, then Feigl reagent,51 the latter reagent removal of the tert-butyl ether group were studied using the tert- showing the presence of a free SI<- group. Although all of the butyl esters of the tert-butoxyamino acids. Two drops of sample reagents except TFA yielded free cysteine, none of the reactions was treated with 3 drops of reagent. After an allotted time the went to completion. The latter reagent gave a product with an reaction was stopped by the addition of an excess of pyridine. Rfvalue of 0.70 which was positive to Feigl reagent. This has The results determined by chromatography of the reaction mix- been tentatively identified as H’cySH .O-t-Bu( L), since a spot ture either on Whatman h-0, 1 paper using the solvent system 1- which was ninhydrin positive with the same Ri value was found in butanol-acetic acid-water, 4: 1:5, (BAW), or on silica gel the startine base (ex. XX’I. plates (thin layer) using the solvent system sec-butyl alcohol- The Rf;slues of the cysteine derivatives in B.4M were deter- 37; ammonia, 3:1 (BAM). termined as: H.c~SH.OH(L), 0.05 H.cy(S-t-Buj .OH(L), Ex. A. Removal of teut-Butyl Groups from H.ser(0-t-Bu) .- 0.19; H.cy(S-t-Bu) ’ O-f-Bu( L), 0.72; H ’C~SH,0-t-Bu, 0.62. -

19.11 Reactions of Aldehydes and Ketones with Amines
19_BRCLoudon_pgs5-0.qxd 12/9/08 11:41 AM Page 926 926 CHAPTER 19 • THE CHEMISTRY OF ALDEHYDES AND KETONES. CARBONYL-ADDITION REACTIONS introduction of the formation of the the protecting group Grignard reagent O S OO HOCH2CH2OH Mg Br C CH3 C p-toluenesulfonic L CH ether LcL L acid (trace) 3 (See Eq. 19.46) Br LcL acetal protecting group; protonolysis of the alkoxide inert to Grignard reagents and hydrolysis of the acetal (removal of the protecting group) O OO OO H2C$ CH2 H2O, H3O| C L C L CH3 L CH3 BrMg BrMg OCH CH LcL |_ 2 2LcL O S HOCH2CH2OH HOCH2CH2 C CH3 (19.54) + LcL L Notice in this synthesis that all steps following acetal formation involve basic or neutral condi- tions. Acid can be used only when destruction of the acetal is desired. Although any acetal group can in principle be used, the five-membered cyclic acetal is fre- quently employed as a protecting group because it forms very rapidly (proximity effect; Sec. 11.7) and it introduces relatively little steric congestion into the protected molecule. A number of reagents that react with carbonyl groups also react with other functional groups. Acetals are commonly used to protect the carbonyl groups of aldehydes and ketones from basic, nucleophilic reagents. Once the protection is no longer needed, the acetal protect- ing group is easily removed, and the carbonyl group re-exposed, by treatment with dilute aqueous acid. Because acetals are unstable in acid, they do not protect carbonyl groups under acidic conditions. PROBLEM 19.27 Outline a synthesis of the following compound from p-bromoacetophenone and any other reagents. -
Iminium and Enamine Catalysis in Enantioselective Photochemical Reactions Cite This: Chem
Chem Soc Rev View Article Online TUTORIAL REVIEW View Journal | View Issue Iminium and enamine catalysis in enantioselective photochemical reactions Cite this: Chem. Soc. Rev., 2018, 47,278 You-Quan Zou, * Fabian M. Ho¨rmann and Thorsten Bach * Although enantioselective catalysis under thermal conditions has been well established over the last few decades, the enantioselective catalysis of photochemical reactions is still a challenging task resulting from the complex enantiotopic face differentiation in the photoexcited state. Recently, remarkable achievements have been reported by a synergistic combination of organocatalysis and photocatalysis, Received 10th August 2017 which have led to the expedient construction of a diverse range of enantioenriched molecules which DOI: 10.1039/c7cs00509a are generally not easily accessible under thermal conditions. In this tutorial review, we summarize and highlight the most significant advances in iminium and enamine catalysis of enantioselective photo- rsc.li/chem-soc-rev chemical reactions, with an emphasis on catalytic modes and reaction types. Creative Commons Attribution-NonCommercial 3.0 Unported Licence. Key learning points (1) The concept of synergistic combination of iminium and enamine catalysis with photocatalysis. (2) The photogeneration of ortho-quinodimethanes, open-shell free radicals and iminium cations. (3) The single-electron-transfer (SET) mechanism in enantioselective photochemical reactions. (4) The direct excitation of iminium ions and enamines in enantioselective photochemical reactions. (5) The electron donor–acceptor (EDA) complex mechanism in enantioselective photochemical reactions. This article is licensed under a 1. Introduction At the dawn of enantioselective photochemistry, circularly polarized light (CPL) was frequently used as the source of Chiral molecules, which can be found in diverse families of chirality.3 However, the use of CPL was limited to specific starting Open Access Article.