Supplement 1 This Supplement Contains the Following Items: 1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Day 2 Day 3 Day 1
Xi’AN Back to the heyday of the Tang Dynasty Location of Xi’an Xi’an is known as Chang’an in ancient times. Having served as the capital of thirteen dynasties, this city is one of the most important places to study and review the history of China. The Tang Dynasty was the pinnacle of China’s history, a period marked by great urbanism and cultural magnificence. As the capital of the Tang Dynasty, Xi’an was the centre of Eastern civilisation. Its importance was comparable to that of Rome in the West. Xi’an’s Tang Dynasty monuments are the most famous of all, and beyond that, the city is committed to recreating the prosperity of the Tang Dynasty. Nowadays, in Xi’an, it is no longer an unattainable dream to travel back in time to the Tang Dynasty. What’s hot Shaanxi History Museum The Shaanxi History Museum is one of the four major museums in China. Its extensive collection of artefacts showcase 1.5 million years of Shaanxi’s history. The third gallery features the culture of the Tang Dynasty as well as artefacts from both the Sui and Tang Dynasties, while the fourth gallery displays a collection of gold and silver artefacts from the Tang Dynasty unearthed in Hejiacun Village. The “Treasure of the Museum”—Agate Cup with Beast’s Head Carving is a superbly crafted jade carving with chic colour. One can also visit the Treasures Gallery and the Tang Dynasty Mural Treasures Gallery of the museum. 91 Xiaozhai East Road, Xiaozhai Commercial Street, Yanta District, Xi’an City, Shaanxi Province, China Take Xi’an Metro Line 1 or 2 and get off at Xiaozhai Station, the museum will be reach from north-east exit. -

China Table of Contents • Dozens Of
Table of Contents Dozens of Bitter Winter Reporters arrested Early Rain pastor accused of inciting subversion Special weekly FORB Newsletter, 21 December 2018 Woman dies after torture during interrogation by Chinese authorities Yu Baorong, a Christian from The Church of Almighty God brutally tortured by Chinese Communist Police Special weekly FORB newsletter, 14 December 2018 ‘I must denounce this wickedness openly’ – detained Chinese pastor Repression of Christian church intensifies China cracks down on Christians -- a new era of religious persecution has arrived Legislative landmark: US Congress passes Reciprocal Access to Tibet Act Yingye’er re-education camp managed like prison (video) 100 church attendees in custody, attacks ongoing Special Weekly FORB Newsletter, 7 December 2018 Government unleashes new round of religious persecution Woman tortured to death by Chinese police: the case of Huang Guorong Woman driven to suicide by the Chinese Communist Government’s long- term harassment: the case of Wang Hongli Special weekly FORB newsletter, 30 November 2018 Uyghurs in China: Position of EU High Representative/Vice-President Mogherini Monetary reward offered for Muslim man’s recapture CCP calls for crackdown against whistleblowers and media Xinjiang authorities sentence Uyghur philanthropist to death for unsanctioned Hajj House church raided twice for standing up to authorities CCTV cameras installed in washrooms at church Burial site forcibly excavated for being “unattractive” (videos) 130 Christians detained -
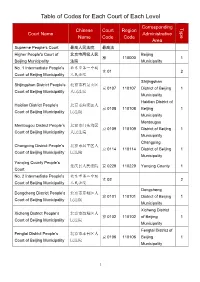
Table of Codes for Each Court of Each Level
Table of Codes for Each Court of Each Level Corresponding Type Chinese Court Region Court Name Administrative Name Code Code Area Supreme People’s Court 最高人民法院 最高法 Higher People's Court of 北京市高级人民 Beijing 京 110000 1 Beijing Municipality 法院 Municipality No. 1 Intermediate People's 北京市第一中级 京 01 2 Court of Beijing Municipality 人民法院 Shijingshan Shijingshan District People’s 北京市石景山区 京 0107 110107 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Haidian District of Haidian District People’s 北京市海淀区人 京 0108 110108 Beijing 1 Court of Beijing Municipality 民法院 Municipality Mentougou Mentougou District People’s 北京市门头沟区 京 0109 110109 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Changping Changping District People’s 北京市昌平区人 京 0114 110114 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Yanqing County People’s 延庆县人民法院 京 0229 110229 Yanqing County 1 Court No. 2 Intermediate People's 北京市第二中级 京 02 2 Court of Beijing Municipality 人民法院 Dongcheng Dongcheng District People’s 北京市东城区人 京 0101 110101 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Xicheng District Xicheng District People’s 北京市西城区人 京 0102 110102 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Fengtai District of Fengtai District People’s 北京市丰台区人 京 0106 110106 Beijing 1 Court of Beijing Municipality 民法院 Municipality 1 Fangshan District Fangshan District People’s 北京市房山区人 京 0111 110111 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Daxing District of Daxing District People’s 北京市大兴区人 京 0115 -

PDF File, 1.4 MB
Annual Report 2020 Contents Important Notes Important Notes 01 1.The Board of Directors (hereinafter referred to as the “Board”), the Supervisory Committee, the directors, the supervisors and senior management of the Bank guarantee the authenticity, accuracy and completeness of the contents of the 2020 Annual Report, in which there are no false representations, misleading statements or material omissions, and are severally Chairman’s Statement 02 and jointly take responsibilities for its contents. Chapter 1 Company Profile 06 2.The 16th meeting of the 11th session of the Board of the Bank deliberated the 2020 Annual Report together with its summary. The meeting required 15 directors to attend, and 15 directors attended the meeting. The Annual Report was approved unanimously at the meeting. Chapter 2 Accounting Data and Financial Indicators 20 3. The 2020 annual financial report prepared by the Bank was audited by PricewaterhouseCoopers Zhong Tian LLP (hereinafter referred to as “PwC”) according to the China Standards on Auditing and PwC issued a standard unqualified auditors’ report. Chapter 3 Discussion and Analysis of Operations 31 4.Xie Yonglin (the Bank’s Chairman), Hu Yuefei (the President), Xiang Youzhi (the Vice President and the CFO) and Zhu Chapter 4 Significant Matters 102 Peiqing (the head of the Finance Department) guarantee the authenticity, accuracy and completeness of the financial reports included in the 2020 Annual Report. Chapter 5 Changes in Shares and Shareholders 118 5.The forward-looking statements such as plans for the future involved in the Report do not constitute a substantial commitment for investors. Investors and stakeholders shall be aware of risks therein and appreciate the distinctions between Chapter 6 Preference Shares 126 plans, forecasts and commitments. -

Research on PM2.5 Concentration Based on Dissipative Structure Theory
www.nature.com/scientificreports OPEN Research on PM2.5 concentration based on dissipative structure theory: a case study of Xi’an, China Xiaoke Sun, Hong Chen*, Zhizhen Liu & Hengrui Chen PM2.5 pollution has become a serious urban environmental problem, especially in developing countries with increasing urbanization. Understanding the proportion of PM2.5 generation sources has laid a foundation for better PM2.5 concentration reduction This paper used Point of Interesting (POI)data, building profle data of Xi’an, PM2.5 concentration and wind monitoring data of fve provinces near Xi’an as the basic data. And this paper studied the spatial distribution of various buildings in Xi’an, the temporal and spatial distribution of PM2.5 in Xi’an and the fve provinces, and found that the spatial distribution of PM2.5 concentration in Xi’an and the building distribution in Xi’an does not match. Based on this, a quantitative model of PM2.5 concentration in Xi’an, energy consumption, wind, and other factors is established through the qualitative and quantitative analysis of PM2.5 concentration in Xi’an. Entropy theory and dissipative structure theory are applied to analyze this phenomenon. The results show PM2.5 in Xi’an mainly comes from the spread of PM2.5 in the fve provinces. The PM2.5 generated by energy consumption in Xi’an is not enough to cause serious PM2.5 pollution. And further suggestions on how to reduce PM2.5 concentration in Xi’an are put forward. With the rapid economic growth and development of infrastructure in developing countries, urbanization and motorization are accelerating. -

Taking Hengyang Central District for Example
E3S Web of Conferences 118, 04022 (2019) https://doi.org/10.1051/e3sconf/201911804022 ICAEER 2019 Study on land use change and its impact on ecosystem service value –Taking Hengyang central district for example Fuqiang Huang1, Zengxiang Qi1,* 1School of Architecture, University of South China, 421001 Hengyang, China Abstract. Land use change on ecosystem service value(ESV) and its interaction is significant in the rapid urbanization. The GIS(Geographic Information System) incorporating with ESV equivalent factor is used to quantitatively study the change of land use change and its impact on the ecosystem service value in Hengyang central district. Further more, the Markov model is used to predict the change of land use and ESV in 2030 under the scenario of historical trend development. The results can serve as a useful tool that assists urban planners in their evaluation of ecosystem service value under the impact of land use change. 1 INTRODUCTION use is often used to predict the quantitative change of land use[9]. It is defined as follows: Ecosystem service is the natural environmental condition of human existence and development formed by S(n) = S(n − 1)P (1) ecosystem in the ecological process[1], Whose’s value can provide scientific basis for human beings to improve Where, S(n) and S(n − 1) is the state of the system at the quality of life and production[2]. Land use the time n and n-1 respectively; is the transition cover/change (LUCC) is the main cause of global probability matrix, which defined as follows: environmental change[3], which plays a decisive role in A A … A the change of ecosystem service. -

Multi-Disciplinary Determination of the Rural/Urban Boundary: a Case Study in Xi’An, China
sustainability Article Multi-Disciplinary Determination of the Rural/Urban Boundary: A Case Study in Xi’an, China Lei Fang 1,2 and Yingjie Wang 1,2,* 1 State Key Lab of Resources and Environmental Information System, Institute of Geographic Sciences and Natural Resources Research, Chinese Academy of Sciences, 11A Datun Road, Chaoyang District, Beijing 100101, China; [email protected] 2 University of Chinese Academy of Sciences, 19A Yuquan Road, Shijingshan District, Beijing 100049, China * Correspondence: [email protected]; Tel.: +86-010-6488-9077 Received: 19 June 2018; Accepted: 23 July 2018; Published: 26 July 2018 Abstract: Rapid urbanization in China has blurred the boundaries between rural and urban areas in both geographic and conceptual terms. Accurately identifying this boundary in a given area is an important prerequisite for studies of these areas, but previous research has used fairly simplistic factors to distinguish the two areas (such as population density). In this study, we built a model combining multi-layer conditions and cumulative percentage methods based on five indicators linking spatial, economic, and demographic factors to produce a more comprehensive and quantitative method for identifying rural and urban areas. Using Xi’an, China as a case study, our methods produced a more accurate determination of the rural-urban divide when compared to data from the National Bureau of Statistics of the People’s Republic of China. Specifically, the urbanization level was 3.24% lower in the new model, with a total urban area that was 621.87 km2 lower. These results were checked by field survey and satellite imagery for accuracy. -

Association of Built Environment with Physical Activity and Health Among Chinese Women Living in Xi'an
Association of built environment with physical activity and health among Chinese women living in Xi'an Yuliang Sun Shaanxi Normal University https://orcid.org/0000-0001-9071-7379 Chunzhen He Shaanxi Normal University Zhiwei Cheng Shaanxi Normal University Jianyu Hou Shaanxi Normal University Fangjun Sun Shaanxi Normal University Xinxin Zhang Shaanxi Normal University Wenfei Zhu ( [email protected] ) https://orcid.org/0000-0002-6660-6350 Research article Keywords: Built Environments, Physical Activity, Health Posted Date: September 24th, 2019 DOI: https://doi.org/10.21203/rs.2.14906/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/13 Abstract Background: The built environment (BE) has been proved to be the factors affecting physical activity(PA) and health level of residents. The aim of this study was to analyze the association of BE with PA and health of Chinese women living in Lianhu District, Xi 'an. Methods: A cross-sectional study was conducted among 202 Chinese women aged 15-69 years old. The International Physical Activity Questionnaire-long version (IPAQ-L) was used to evaluate PA. BE was measured by the Neighborhood Environment Walkability Scale - Abbreviated (NEWS-A). General linear regression models were created to assess the relationship among BE, PA and health related variables, including body mass index (BMI), body fat percentage (BFP), systolic blood pressure (SBP), and diastolic blood pressure(DBP). All the models were controlled for age. Results: Transportation PA were negatively associated with residential density( P <0.05). No signicant correlation was found between BE and leisure time PA( P >0.05). -

Minimum Wage Standards in China August 11, 2020
Minimum Wage Standards in China August 11, 2020 Contents Heilongjiang ................................................................................................................................................. 3 Jilin ............................................................................................................................................................... 3 Liaoning ........................................................................................................................................................ 4 Inner Mongolia Autonomous Region ........................................................................................................... 7 Beijing......................................................................................................................................................... 10 Hebei ........................................................................................................................................................... 11 Henan .......................................................................................................................................................... 13 Shandong .................................................................................................................................................... 14 Shanxi ......................................................................................................................................................... 16 Shaanxi ...................................................................................................................................................... -

Evaluation of the Equity and Regional Management of Some Urban Green Space Ecosystem Services: a Case Study of Main Urban Area of Xi’An City
Article Evaluation of the Equity and Regional Management of Some Urban Green Space Ecosystem Services: A Case Study of Main Urban Area of Xi’an City Hui Dang 1, Jing Li 1,*, Yumeng Zhang 1 and Zixiang Zhou 2 1 School of Geography and Tourism, Shaanxi Normal University, Xi’an 710119, China; [email protected] (H.D.); [email protected] (Y.Z.) 2 College of Geomatics, Xi’an University of Science and Technology, Xi’an 710054, China; [email protected] * Correspondence: [email protected]; Tel.: +86-137-2061-8191 Abstract: Urban green spaces can provide many types of ecosystem services for residents. An imbalance in the pattern of green spaces leads to an inequality of the benefits of such spaces. Given the current situation of environmental problems and the basic geographical conditions of Xi’an City, this study evaluated and mapped four kinds of ecosystem services from the perspective of equity: biodiversity, carbon sequestration, air purification, and climate regulation. Regionalization with dynamically constrained agglomerative clustering and partitioning (REDCAP) was used to obtain the partition groups of ecosystem services. The results indicate that first, the complexity of the urban green space community is low, and the level of biodiversity needs to be improved. The dry deposition flux of particulate matter (PM2.5) decreases from north to south, and green spaces enhance the adsorption of PM2.5. Carbon sequestration in the south and east is higher than that in the north and west, respectively. The average surface temperature in green spaces is lower than that in other urban areas. -

Study on Temporal and Spatial Variation Characteristics and Influencing Factors of Land Use Efficiency in Xi’An, China
sustainability Article Study on Temporal and Spatial Variation Characteristics and Influencing Factors of Land Use Efficiency in Xi’an, China Jing Huang and Dongqian Xue * School of Geographical Science and Tourism, Shaanxi Normal University, Xi’an 710062, China; [email protected] * Correspondence: [email protected] Received: 17 October 2019; Accepted: 22 November 2019; Published: 25 November 2019 Abstract: China’s urban land use has shifted from incremental expansion to inventory eradication. The traditional extensive management mode is difficult to maintain, and the fundamental solution is to improve land use efficiency. Xi’an, the largest central city in Western China, was selected as the research area. The super-efficiency data envelopment analysis (DEA) model and Malmquist index method were used to measure the land use efficiency of each district and county in the city from the micro perspective, and the spatial-temporal change characteristics and main influencing factors of land use efficiency were analyzed, which not only made up for the research content of urban land use efficiency in China’s underdeveloped areas, but also pointed out the emphasis and direction for the improvement of urban land use efficiency. The results showed that: (1) The land use efficiency of Xi’an reflected the land use intensive level of the underdeveloped areas in Western China, that is, the overall intensive level was not high, the gap between the urban internal land use efficiency was large, the land use efficiency of the old urban area and the mature built-up area was relatively high, and the land use efficiency of the emerging expansion area and the edge area was relatively low. -

Shanxi WLAN Hotspots 1/23
Shanxi WLAN hotspots NO. SSID Location_Name Location_Type Location_Address City Province 1 ChinaNet Xi'an Beilin Qu friendship distance tea lounge bar entertainment bars Friendship Road No. 260 on the 1st Deputy Xi'An shaanxi 2 ChinaNet Xi'an Aidi Food Management Co., Ltd. entertainment bars Friendship Road 127, creativity Xi'An shaanxi 3 ChinaNet Xi'an Lianhu District Run-Xuan Teahouse entertainment bars Friendship Road 127, 53, deputy Xi'An shaanxi 4 ChinaNet Yanta District in Xi'an Ziwei Wang waterfall poor artists entertainment bars E-249, all the way Xi'An shaanxi Xi'an high-tech zones love Qindao Art of Living Museum 5 ChinaNet entertainment bars E-foot Street North, 103 East Street, I love Qindao cafe Xi'An shaanxi coffee bad language 6 ChinaNet Yanta District in Xi'an Ming Shi-language music coffee tea entertainment bars Han Guang Ming-shi, 100 South Lok coffee language tea Xi'An shaanxi 7 ChinaNet Xi'an Lianhu District Duoji Cafe entertainment bars Labor South on the 17th to pay on the 22nd on the 23rd Xi'An shaanxi 8 ChinaNet Xian Ou goods Food Co., Ltd. entertainment bars Road work on the 17th to pay on the 12th Xi'An shaanxi 9 ChinaNet Henrik Business Hotel hotels Auspicious Road Xi'An shaanxi 10 ChinaNet Xian Ming Jue Dining Entertainment Co., Ltd. entertainment bars High road on the 23rd Xi'An shaanxi 11 ChinaNet Tang Yan Tang Ruins Park Plaza Road entertainment bars Tang Yan Road Xi'An shaanxi 12 ChinaNet HSBC coffee entertainment bars Technology Road Xi'An shaanxi 13 ChinaNet Xi'an high-tech love Investment Group Co., Ltd.