Transcriptional Profiling of Cardiac Cells Links Age-Dependent
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

PLXNB1 (Plexin
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL AT INIST-CNRS Gene Section Mini Review PLXNB1 (plexin B1) José Javier Gómez-Román, Montserrat Nicolas Martínez, Servando Lazuén Fernández, José Fernando Val-Bernal Department of Anatomical Pathology, Marques de Valdecilla University Hospital, Medical Faculty, University of Cantabria, Santander, Spain (JJGR, MN, SL, JFVB) Published in Atlas Database: March 2009 Online updated version: http://AtlasGeneticsOncology.org/Genes/PLXNB1ID43413ch3p21.html DOI: 10.4267/2042/44702 This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2010 Atlas of Genetics and Cytogenetics in Oncology and Haematology Identity Pseudogene No. Other names: KIAA0407; MGC149167; OTTHUMP00000164806; PLEXIN-B1; PLXN5; SEP Protein HGNC (Hugo): PLXNB1 Location: 3p21.31 Description Local order: The Plexin B1 gene is located between 2135 Amino acids (AA). Plexins are receptors for axon FBXW12 and CCDC51 genes. molecular guidance molecules semaphorins. Plexin signalling is important in pathfinding and patterning of both neurons and developing blood vessels. Plexin-B1 is a surface cell receptor. When it binds to its ligand SEMA4D it activates several pathways by binding of cytoplasmic ligands, like RHOA activation and subsequent changes of the actin cytoskeleton, axon guidance, invasive growth and cell migration. It monomers and heterodimers with PLXNB2 after proteolytic processing. Binds RAC1 that has been activated by GTP binding. It binds PLXNA1 and by similarity ARHGEF11, Note ARHGEF12, ERBB2, MET, MST1R, RND1, NRP1 Size: 26,200 bases. and NRP2. Orientation: minus strand. This family features the C-terminal regions of various plexins. The cytoplasmic region, which has been called DNA/RNA a SEX domain in some members of this family is involved in downstream signalling pathways, by Description interaction with proteins such as Rac1, RhoD, Rnd1 and other plexins. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -
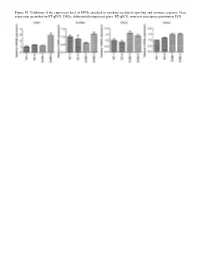
Figure S1. Validation of the Expression Level of Degs Enriched in Cytokine‑Mediated Signaling and Immune Response
Figure S1. Validation of the expression level of DEGs enriched in cytokine‑mediated signaling and immune response. Gene expression quantified by RT‑qPCR. DEGs, differentially expressed genes; RT‑qPCR, reverse‑transcription quantitative PCR. Figure S2. Validation of STAU1‑regulated AS events. IGV‑Sashimi plot revealed (A‑C) three A5SS AS events in three different genes. Reads distribution of each AS event was plotted in the left panel with the transcripts of each gene shown below. The sche‑ matic diagrams depict the structures of two AS events, AS1 (purple line) and AS2 (green line). The exon sequences are denoted by black boxes, the intron sequences by a horizontal line (right panel). RNA‑seq quantification and RT‑qPCR validation of ASEs are presented in the panels on the right. STAU1, double‑stranded RNA‑binding protein Staufen homolog 1; AS, alternative splicing; A5SS, alternative 5'splice site; RNA‑seq, RNA sequencing; RT‑qPCR, reverse‑transcription quantitative PCR. Error bars represent mean ± SEM. *P<0.05. Table SI. Primers used in gene validation experiments. IFIT2‑F CAGCCTACGGCAACTAAA IFIT2‑R GAGCCTTCTCAAAGCACA IFIT3‑F ACACCAAACAATGGCTAC IFIT3‑R TGGACAAACCCTCTAAAC OASL‑F AATGGTGACCGTGATGGG OASL‑R ACCTGAGGATGGAGCAGAG IFI27‑F TTCACTGCGGCGGGAATC IFI27‑R TGGCTGCTATGGAGGACGAG S1PR4‑F TGCTGAAGACGGTGCTGATG S1PR4‑R TGCGGAAGGAGTAGATGATGG CCL5‑F ACGACTGCTGGGTTGGAG CCL5‑R ACCCTGCTGCTTTGCCTA CCL2‑F CTAACCCAGAAACATCCAAT CCL2‑R GCTATGAGCAGCAGGCAC CD44‑F TGGAGGACAGAAAGCCAAGT CD44‑R TTCGCAATGAAACAATCAGTAG PLEKHG2‑M/As‑F CCAAAAGTAAGCCTGTCC PLEKHG2‑M‑R -

The KMT1A-GATA3-STAT3 Circuit Is a Novel Self-Renewal Signaling of Human Bladder Cancer Stem Cells Zhao Yang1, Luyun He2,3, Kais
The KMT1A-GATA3-STAT3 circuit is a novel self-renewal signaling of human bladder cancer stem cells Zhao Yang1, Luyun He2,3, Kaisu Lin4, Yun Zhang1, Aihua Deng1, Yong Liang1, Chong Li2, 5, & Tingyi Wen1, 6, 1CAS Key Laboratory of Pathogenic Microbiology and Immunology, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China 2Core Facility for Protein Research, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China 3CAS Key Laboratory of Infection and Immunity, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China 4Department of Oncology, the Second Affiliated Hospital of Soochow University, Suzhou 215000, China 5Beijing Jianlan Institute of Medicine, Beijing 100190, China 6Savaid Medical School, University of Chinese Academy of Sciences, Beijing 100049, China Correspondence author: Tingyi Wen, e-mail: [email protected] Chong Li, e-mail: [email protected] Supplementary Figure S1. Isolation of human bladder cancer stem cells. BCMab1 and CD44 were used to isolate bladder cancer stem cells (BCSCs: BCMab1+CD44+) and bladder cancer non-stem cells (BCNSCs: BCMab1-CD44-) from EJ, samples #1 and #2 by flow cytometry. Supplementary Figure S2. Gene ontology analysis of downregulated genes of human BCSCs. (A) Pathway enrichment of 103 downregulated genes in BCSCs. (B) The seven downregulated genes in BCSCs participating in centromeric heterochromatin, mRNA-3’-UTR binding and translation regulator activity signaling pathways were validated by qRT-PCR. Data are presented as mean ± SD. P < 0.05; P < 0.01. Supplementary Figure S3. The expression of KMT1A is higher in human BC than that in peri-tumor tissues. (A) The expression of KMT1A was higher in BC samples than that in peri-tumors as assessed by immunohistochemistry, Scale bar = 50 m. -

Biological Models of Colorectal Cancer Metastasis and Tumor Suppression
BIOLOGICAL MODELS OF COLORECTAL CANCER METASTASIS AND TUMOR SUPPRESSION PROVIDE MECHANISTIC INSIGHTS TO GUIDE PERSONALIZED CARE OF THE COLORECTAL CANCER PATIENT By Jesse Joshua Smith Dissertation Submitted to the Faculty of the Graduate School of Vanderbilt University In partial fulfillment of the requirements For the degree of DOCTOR OF PHILOSOPHY In Cell and Developmental Biology May, 2010 Nashville, Tennessee Approved: Professor R. Daniel Beauchamp Professor Robert J. Coffey Professor Mark deCaestecker Professor Ethan Lee Professor Steven K. Hanks Copyright 2010 by Jesse Joshua Smith All Rights Reserved To my grandparents, Gladys and A.L. Lyth and Juanda Ruth and J.E. Smith, fully supportive and never in doubt. To my amazing and enduring parents, Rebecca Lyth and Jesse E. Smith, Jr., always there for me. .my sure foundation. To Jeannine, Bill and Reagan for encouragement, patience, love, trust and a solid backing. To Granny George and Shawn for loving support and care. And To my beautiful wife, Kelly, My heart, soul and great love, Infinitely supportive, patient and graceful. ii ACKNOWLEDGEMENTS This work would not have been possible without the financial support of the Vanderbilt Medical Scientist Training Program through the Clinical and Translational Science Award (Clinical Investigator Track), the Society of University Surgeons-Ethicon Scholarship Fund and the Surgical Oncology T32 grant and the Vanderbilt Medical Center Section of Surgical Sciences and the Department of Surgical Oncology. I am especially indebted to Drs. R. Daniel Beauchamp, Chairman of the Section of Surgical Sciences, Dr. James R. Goldenring, Vice Chairman of Research of the Department of Surgery, Dr. Naji N. -

WO 2012/174282 A2 20 December 2012 (20.12.2012) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2012/174282 A2 20 December 2012 (20.12.2012) P O P C T (51) International Patent Classification: David [US/US]; 13539 N . 95th Way, Scottsdale, AZ C12Q 1/68 (2006.01) 85260 (US). (21) International Application Number: (74) Agent: AKHAVAN, Ramin; Caris Science, Inc., 6655 N . PCT/US20 12/0425 19 Macarthur Blvd., Irving, TX 75039 (US). (22) International Filing Date: (81) Designated States (unless otherwise indicated, for every 14 June 2012 (14.06.2012) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, English (25) Filing Language: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, Publication Language: English DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, (30) Priority Data: KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, 61/497,895 16 June 201 1 (16.06.201 1) US MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, 61/499,138 20 June 201 1 (20.06.201 1) US OM, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SC, SD, 61/501,680 27 June 201 1 (27.06.201 1) u s SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, 61/506,019 8 July 201 1(08.07.201 1) u s TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

WNT16 Is a New Marker of Senescence
Table S1. A. Complete list of 177 genes overexpressed in replicative senescence Value Gene Description UniGene RefSeq 2.440 WNT16 wingless-type MMTV integration site family, member 16 (WNT16), transcript variant 2, mRNA. Hs.272375 NM_016087 2.355 MMP10 matrix metallopeptidase 10 (stromelysin 2) (MMP10), mRNA. Hs.2258 NM_002425 2.344 MMP3 matrix metallopeptidase 3 (stromelysin 1, progelatinase) (MMP3), mRNA. Hs.375129 NM_002422 2.300 HIST1H2AC Histone cluster 1, H2ac Hs.484950 2.134 CLDN1 claudin 1 (CLDN1), mRNA. Hs.439060 NM_021101 2.119 TSPAN13 tetraspanin 13 (TSPAN13), mRNA. Hs.364544 NM_014399 2.112 HIST2H2BE histone cluster 2, H2be (HIST2H2BE), mRNA. Hs.2178 NM_003528 2.070 HIST2H2BE histone cluster 2, H2be (HIST2H2BE), mRNA. Hs.2178 NM_003528 2.026 DCBLD2 discoidin, CUB and LCCL domain containing 2 (DCBLD2), mRNA. Hs.203691 NM_080927 2.007 SERPINB2 serpin peptidase inhibitor, clade B (ovalbumin), member 2 (SERPINB2), mRNA. Hs.594481 NM_002575 2.004 HIST2H2BE histone cluster 2, H2be (HIST2H2BE), mRNA. Hs.2178 NM_003528 1.989 OBFC2A Oligonucleotide/oligosaccharide-binding fold containing 2A Hs.591610 1.962 HIST2H2BE histone cluster 2, H2be (HIST2H2BE), mRNA. Hs.2178 NM_003528 1.947 PLCB4 phospholipase C, beta 4 (PLCB4), transcript variant 2, mRNA. Hs.472101 NM_182797 1.934 PLCB4 phospholipase C, beta 4 (PLCB4), transcript variant 1, mRNA. Hs.472101 NM_000933 1.933 KRTAP1-5 keratin associated protein 1-5 (KRTAP1-5), mRNA. Hs.534499 NM_031957 1.894 HIST2H2BE histone cluster 2, H2be (HIST2H2BE), mRNA. Hs.2178 NM_003528 1.884 CYTL1 cytokine-like 1 (CYTL1), mRNA. Hs.13872 NM_018659 tumor necrosis factor receptor superfamily, member 10d, decoy with truncated death domain (TNFRSF10D), 1.848 TNFRSF10D Hs.213467 NM_003840 mRNA. -

Suppl Fig 2 PB2 EUCOMM V3
Maier et al. Supplementary material Semaphorin 4C and 4G are Plexin-B2 ligands required in cerebellar development 2 Supplementary tables 4 Supplementary figures 1 Supplementary Table 1. Exencephaly and postnatal survival of Sema4C mutants (on C57BL/6 background) Genotype Expected frequency Observed frequency Exencephaly Embryonic stages (n=47)* Wild type 25% 25% (12/47) 0% Sema4c+/- 50% 45% (21/47) 0% Sema4c-/- 25% 30% (14/47) 36% (5/14) Postnatal animals (n=417)* Wild type 25% 27% (113/417) Sema4c+/- 50% 61% (253/417) Sema4c-/- 25% 12% (51/417) * Offspring from Sema4c+/- x +/- matings. Embryonic stages scored at E15-E18, postnatal animals scored at P21. 2 Supplementary Table 2. Cerebellar phenotypes of Sema4C and Sema4G mutants (on CD-1 outbred background) Ectopic Fusion of Gap in Normal Gap in IGL granule cells lobules IGL of cerebellum of lobule II in molecular VIII/IX* lobule X Genotype n layer weak strong Wild type 17 100% 0% 0% 0% 0% 0% Sema4c+/- 11 70% 10% 0% 10% 0% 10% Sema4c-/- 10 20% 70% 0% 40% 0% 40% Sema4c+/-; Sema4g+/- 16 55% 18% 0% 18% 0% 18% Sema4c+/-; Sema4g-/- 13 40% 20% 0% 20% 0% 40% Sema4c-/-; Sema4g+/- 10 20% 30% 0% 50% 0% 60% Sema4c-/-; Sema4g-/- 11 0% 55% 0% 55% 55% 82% *Fusions of lobules VIII and IX were scored as “weak” when ectopic granule cells formed a band of cells at the fusion line, and as “strong” when a continuous bridge of granule cells connected the IGL of the two lobules. 3 Supplementary figure legends Maier et al. Suppl. Fig. -

The Role of Natural Selection in Human Evolution – Insights from Latin America
Genetics and Molecular Biology, 39, 3, 302-311 (2016) Copyright © 2016, Sociedade Brasileira de Genética. Printed in Brazil DOI: http://dx.doi.org/10.1590/1678-4685-GMB-2016-0020 Review Article The role of natural selection in human evolution – insights from Latin America Francisco M. Salzano1 1Departamento de Genética, Instituto de Biociências, Universidade Federal do Rio Grande do Sul (UFRGS), Porto Alegre, RS, Brazil. Abstract A brief introduction considering Darwin’s work, the evolutionary synthesis, and the scientific biological field around the 1970s and subsequently, with the molecular revolution, was followed by selected examples of recent investiga- tions dealing with the selection-drift controversy. The studies surveyed included the comparison between essential genes in humans and mice, selection in Africa and Europe, and the possible reasons why females in humans remain healthy and productive after menopause, in contrast with what happens in the great apes. At the end, selected exam- ples of investigations performed in Latin America, related to the action of selection for muscle performance, acetylation of xenobiotics, high altitude and tropical forest adaptations were considered. Despite dissenting views, the influence of positive selection in a considerable portion of the human genome cannot presently be dismissed. Keywords: natural selection, human evolution, population genetics, human adaptation, history of genetics. Received: February 10, 2016; Accepted: May 8, 2016. History deniably indicated that we had derived from -

A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2018 A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family Linda Molla Follow this and additional works at: https://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Linda Molla June 2018 © Copyright by Linda Molla 2018 A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY Linda Molla, Ph.D. The Rockefeller University 2018 APOBEC2 is a member of the AID/APOBEC cytidine deaminase family of proteins. Unlike most of AID/APOBEC, however, APOBEC2’s function remains elusive. Previous research has implicated APOBEC2 in diverse organisms and cellular processes such as muscle biology (in Mus musculus), regeneration (in Danio rerio), and development (in Xenopus laevis). APOBEC2 has also been implicated in cancer. However the enzymatic activity, substrate or physiological target(s) of APOBEC2 are unknown. For this thesis, I have combined Next Generation Sequencing (NGS) techniques with state-of-the-art molecular biology to determine the physiological targets of APOBEC2. Using a cell culture muscle differentiation system, and RNA sequencing (RNA-Seq) by polyA capture, I demonstrated that unlike the AID/APOBEC family member APOBEC1, APOBEC2 is not an RNA editor. Using the same system combined with enhanced Reduced Representation Bisulfite Sequencing (eRRBS) analyses I showed that, unlike the AID/APOBEC family member AID, APOBEC2 does not act as a 5-methyl-C deaminase. -

Combinatorial Strategies Using CRISPR/Cas9 for Gene Mutagenesis in Adult Mice
Combinatorial strategies using CRISPR/Cas9 for gene mutagenesis in adult mice Avery C. Hunker A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2019 Reading Committee: Larry S. Zweifel, Chair Sheri J. Mizumori G. Stanley McKnight Program Authorized to Offer Degree: Pharmacology 2 © Copyright 2019 Avery C. Hunker 3 University of Washington ABSTRACT Combinatorial strategies using CRISPR/Cas9 for gene mutagenesis in adult mice Avery C. Hunker Chair of the Supervisory Committee: Larry Zweifel Department of Pharmacology A major challenge to understanding how genes modulate complex behaviors is the inability to restrict genetic manipulations to defined cell populations or circuits. To circumvent this, we created a simple strategy for limiting gene knockout to specific cell populations using a viral-mediated, conditional CRISPR/SaCas9 system in combination with intersectional genetic strategies. A small single guide RNA (sgRNA) directs Staphylococcus aureus CRISPR-associated protein (SaCas9) to unique sites on DNA in a Cre-dependent manner resulting in double strand breaks and gene mutagenesis in vivo. To validate this technique we targeted nine different genes of diverse function in distinct cell types in mice and performed an array of analyses to confirm gene mutagenesis and subsequent protein loss, including IHC, cell-type specific DNA sequencing, electrophysiology, Western blots, and behavior. We show that these vectors are as efficient as conventional conditional gene knockout and provide a viable alternative to complex genetic crosses. This strategy provides additional benefits of 4 targeting gene mutagenesis to cell types previously difficult to isolate, and the ability to target genes in specific neural projections for gene inactivation.