Synthesis and Evaluation of Small Molecule Inhibitors of Pyruvate Carboxylase
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Construction and Enzymatic Characterization of E
1 Minimizing acetate formation in E. coli fermentations 2 Marjan De Mey*, Sofie De Maeseneire, Wim Soetaert and Erick Vandamme 3 Ghent University, Laboratory of Industrial Microbiology and Biocatalysis, Department of 4 Biochemical and Microbial Technology, Faculty of Bioscience Engineering, Coupure links 5 653, B-9000 Ghent, Belgium 6 7 * Correspondence address: Marjan De Mey 8 Laboratory of Industrial Microbiology and Biocatalysis 9 Department of Biochemical and Microbial Technology 10 Faculty of Bioscience Engineering 11 Ghent University 12 Coupure links 653 13 B-9000 Gent 14 Tel: +32-9-264-60-28 15 Fax: +32-9-264-62-31 16 [email protected] 17 Keywords: acetate reduction, Escherichia coli, metabolic engineering 1 18 Abstract 19 Escherichia coli remains the best established production organisms in industrial 20 biotechnology. However, during aerobic fermentation runs at high growth rates, considerable 21 amounts of acetate are accumulated as by-product. This by-product has negative effects on 22 growth and protein production. Over the last 20 years, substantial research efforts have been 23 spent to reduce acetate accumulation during aerobic growth of E. coli on glucose. From the 24 onset it was clear that this quest should not be a simple nor uncomplicated one. Simple 25 deletion of the acetate pathway, reduced the acetate accumulation, but instead other by- 26 products were formed. This minireview gives a clear outline of these research efforts and the 27 outcome of them, including bioprocess level approaches and genetic approaches. Recently, 28 the latter seems to have some promising results. 29 30 1 Introduction 31 Escherichia coli was the first and is still one of the most commonly used production 32 organisms in industrial biotechnology. -

UMPOLUNG in REACTIONS CATALYZED by THIAMINE PYROPHOSPHATE DEPENDENT ENZYMES Umpolung En Reacciones Catalizadas Por Enzimas Dependientes De Pirofosfato De Tiamina
Ciencia, Ambiente y Clima, Vol. 2, No. 2, julio-diciembre, 2019 • ISSN (impreso): 2636-2317 • ISSN (en línea): 2636-2333 DOI: https://doi.org/10.22206/cac.2019.v2i2.pp27-42 UMPOLUNG IN REACTIONS CATALYZED BY THIAMINE PYROPHOSPHATE DEPENDENT ENZYMES Umpolung en reacciones catalizadas por enzimas dependientes de pirofosfato de tiamina Carlos José Boluda Emily Soto Instituto Tecnológico de Santo Domingo (INTEC), Instituto Tecnológico de Santo Domingo (INTEC), Área de Ciencias Básicas y Ambientales, Av. de Los Área de Ciencias Básicas y Ambientales Próceres 49, Santo Domingo, República Dominicana Correo-e: [email protected] *Corresponding author: Carlos J. Boluda Darah de la Cruz Correo-e: [email protected] Instituto Tecnológico de Santo Domingo (INTEC), Carolina Juncá Área de Ciencias Básicas y Ambientales Instituto Tecnológico de Santo Domingo (INTEC), Correo-e: [email protected] Área de Ciencias Básicas y Ambientales Anny Peña Correo-e: [email protected] Instituto Tecnológico de Santo Domingo (INTEC), Área de Ciencias Básicas y Ambientales Correo-e: [email protected] Recibido: 25/9/2019 • Aprobado: 19/10/2019 Cómo citar: Boluda, C. J., Juncá, C., Soto, E., de la Cruz, D., & Peña, A. (2019). Umpolung in reactions catalyzed by thiamine pyrophos- phate dependent enzymes. Ciencia, Ambiente Y Clima, 2(2), 27-42. Doi: https://doi.org/10.22206/cac.2019.v2i2.pp27-42 Abstract Resumen The temporal exchange of the electrophilic/nucleophilic El intercambio temporal del carácter electrofílico/nucleofí- character of an atom by chemical manipulation is known lico de un átomo mediante manipulación química, es cono- in organic chemistry as umpolung. This inversion of polarity cido con el vocablo alemán de umpolung. -

Targeting Pyruvate Carboxylase Reduces Gluconeogenesis and Adiposity and Improves Insulin Resistance Naoki Kumashiro,1,2 Sara A
ORIGINAL ARTICLE Targeting Pyruvate Carboxylase Reduces Gluconeogenesis and Adiposity and Improves Insulin Resistance Naoki Kumashiro,1,2 Sara A. Beddow,3 Daniel F. Vatner,2 Sachin K. Majumdar,2 Jennifer L. Cantley,1,2 Fitsum Guebre-Egziabher,2 Ioana Fat,2 Blas Guigni,2 Michael J. Jurczak,2 Andreas L. Birkenfeld,2 Mario Kahn,2 Bryce K. Perler,2 Michelle A. Puchowicz,4 Vara Prasad Manchem,5 Sanjay Bhanot,5 Christopher D. Still,6 Glenn S. Gerhard,6 Kitt Falk Petersen,2 Gary W. Cline,2 Gerald I. Shulman,1,2,7 and Varman T. Samuel2,3 – We measured the mRNA and protein expression of the key transcriptional factors and cofactors (8 12). Yet, despite gluconeogenic enzymes in human liver biopsy specimens and the high degree of transcription regulation for these found that only hepatic pyruvate carboxylase protein levels enzymes, the control they exert over gluconeogenic flux is related strongly with glycemia. We assessed the role of pyruvate relatively weak (13–16). We recently reported that hepatic carboxylase in regulating glucose and lipid metabolism in rats expression of PEPCK and G6PC mRNA was not related to through a loss-of-function approach using a specific antisense fasting hyperglycemia in two rodent models of type 2 di- oligonucleotide (ASO) to decrease expression predominantly in abetes and in patients with type 2 diabetes (17). Thus, we liver and adipose tissue. Pyruvate carboxylase ASO reduced hypothesized that other mechanisms must account for in- plasma glucose concentrations and the rate of endogenous glucose production in vivo. Interestingly, pyruvate carboxylase ASO also creased hepatic gluconeogenesis and fasting hyperglycemia reduced adiposity, plasma lipid concentrations, and hepatic in type 2 diabetes. -
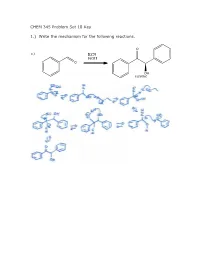
CHEM 345 Problem Set 18 Key 1.) Write the Mechanism for The
CHEM 345 Problem Set 18 Key 1.) Write the mechanism for the following reactions. O a.) KCN EtOH O OH racemic 1.) Write the mechanism for the following reactions. b.) KCN AcOH O NC OH racemic O c.) N S R O NEt3 OH racemic 2.) What is the structure of AcOH?Why does changing the solvent from EtOH to AcOH make such a big difference? O OH AcOH acetic acid The pKa of acetic acid is approximately 5. The pKa of ethanol is approximately 15. When you take a proton off of ethanol, you generate ethoxide which is about 1010 times stronger of a base than acetate. 3.) Give two instances when you need to use the thiazolium salt and triethylamine rather than KCN and EtOH. If the aldehydes contain an enolizable proton then you cannot use KCN/EtOH, instead you must use the thiazolium. Also, if the electrophile is a Michael acceptor to give a 1,4 dicarbonyl, then the thiazolium catalyst should be used. 4.) Break the following compound down as far as you can using Aldol, Michael, and Claisen reactions. Above each retrosynthetic arrow, write the name of the reaction. O HO O HO Aldol O Michael HO O O HO O Aldol O O Aldol HO O O HO Michael O O O Aldol There are other possibilities for order. O O HO Aldol O O 5.) Synthesize the following molecules. All carbons in the molecules must come from benzene or compounds with 5C’s or less. a.) O H2SO4 O HNO3 O2N AlCl3 O O SOCl2 HO Cl H2CrO4 1.) BuLi, Et2O + O 2.) H3O HO b.) O O O Cl + H3O NaOEt, EtOH O O O O Cl O AlCl3 Cl Cl AlCl3 Cl2 c.) O OMe NaOMe MeOH O O 1.) POCl3, DMF 2.) H2O OMe OMe O AlCl3 MeI Cl ONa HCl NaOH ZnHg 1.) NaOH + mcpba O 2.) H3O O OH O O AlCl3 Cl 6.) Write the mechanism for the following reactions. -

Citric Acid Cycle
CHEM464 / Medh, J.D. The Citric Acid Cycle Citric Acid Cycle: Central Role in Catabolism • Stage II of catabolism involves the conversion of carbohydrates, fats and aminoacids into acetylCoA • In aerobic organisms, citric acid cycle makes up the final stage of catabolism when acetyl CoA is completely oxidized to CO2. • Also called Krebs cycle or tricarboxylic acid (TCA) cycle. • It is a central integrative pathway that harvests chemical energy from biological fuel in the form of electrons in NADH and FADH2 (oxidation is loss of electrons). • NADH and FADH2 transfer electrons via the electron transport chain to final electron acceptor, O2, to form H2O. Entry of Pyruvate into the TCA cycle • Pyruvate is formed in the cytosol as a product of glycolysis • For entry into the TCA cycle, it has to be converted to Acetyl CoA. • Oxidation of pyruvate to acetyl CoA is catalyzed by the pyruvate dehydrogenase complex in the mitochondria • Mitochondria consist of inner and outer membranes and the matrix • Enzymes of the PDH complex and the TCA cycle (except succinate dehydrogenase) are in the matrix • Pyruvate translocase is an antiporter present in the inner mitochondrial membrane that allows entry of a molecule of pyruvate in exchange for a hydroxide ion. 1 CHEM464 / Medh, J.D. The Citric Acid Cycle The Pyruvate Dehydrogenase (PDH) complex • The PDH complex consists of 3 enzymes. They are: pyruvate dehydrogenase (E1), Dihydrolipoyl transacetylase (E2) and dihydrolipoyl dehydrogenase (E3). • It has 5 cofactors: CoASH, NAD+, lipoamide, TPP and FAD. CoASH and NAD+ participate stoichiometrically in the reaction, the other 3 cofactors have catalytic functions. -

Mechanism F Formation of Oxaloacetate and Phosphoenol Pyruvate from Pyruvate1
THE JOURNAL OF VITAMINOLOGY 14, 59-67 (1968) MECHANISM F FORMATION OF OXALOACETATE AND PHOSPHOENOL PYRUVATE FROM PYRUVATE1 HARLAND G. WOOD Department of Biochemistry, Case Western Reserve University Cleveland, Ohio 44, 106, U.S.A. Pyruvate, oxaloacetate and P-enolpyruvate have been known to be key compounds in metabolism for many years; pyruvate since 1911 when Neuberg found that yeast decarboxylates pyruvate to acetaldehyde, oxaloacetate since 1937 when Krebs pro posed the citric acid cycle, and P-enolpyruvate since 1934-1936 when Lohmann, Meyerhof and Kiessling discovered that 3-phosphoglycerate is converted to P enolpyruvate by dialyzed yeast or muscle extracts. These three compounds are the hub of numerous reactions leading to both synthesis and breakdown of carbo hydrates, fats and nitrogenous compounds (See the recent review by Utter (1) ). Furthermore, they are interlocked within themselves by a series of reactions re sulting in the synthesis of each fromn the other with accompanying breakdown of the companion compound. There has been a long debate concerning the mechanism of formation of P enolpyruvate from pyruvate. The conversion of P-enolpyruvate to pyruvate in glycolysis by pyruvate kinase was shown by Lardy and Ziegler (2) in 1945 to be reversible, but the equilibrium lies far to the right and the physiological signi ficance of the reversibility is questionable. The possibility that P-enolpyruvate might be formed by a pathway other than via pyruvate kinase was first raised by Kalckar (3) who found that kidney preparations could form P-enolpyruvate from malate and fumarate. At that time Wood and Werkman had discovered fixation of CO2 by heterotrophs and had postulated that the fixation occurs by combination of CO2 with pyruvate to give oxaloacetate. -

Pyruvate Carboxylase Signalling Axis Couples Mitochondrial Metabolism to Glucose-Stimulated Insulin Secretion in Pancreatic B-Cells
ARTICLE Received 23 Oct 2015 | Accepted 26 Apr 2016 | Published 6 Jun 2016 DOI: 10.1038/ncomms11740 OPEN The MDM2–p53–pyruvate carboxylase signalling axis couples mitochondrial metabolism to glucose-stimulated insulin secretion in pancreatic b-cells Xiaomu Li1,2,3,*, Kenneth K.Y. Cheng1,2,*, Zhuohao Liu1,2, Jin-Kui Yang4, Baile Wang1,2, Xue Jiang1,2, Yawen Zhou1,2, Philip Hallenborg5, Ruby L.C. Hoo1,2, Karen S.L. Lam1,2, Yasuhiro Ikeda6, Xin Gao3 & Aimin Xu1,2,7 Mitochondrial metabolism is pivotal for glucose-stimulated insulin secretion (GSIS) in pancreatic b-cells. However, little is known about the molecular machinery that controls the homeostasis of intermediary metabolites in mitochondria. Here we show that the activation of p53 in b-cells, by genetic deletion or pharmacological inhibition of its negative regulator MDM2, impairs GSIS, leading to glucose intolerance in mice. Mechanistically, p53 activation represses the expression of the mitochondrial enzyme pyruvate carboxylase (PC), resulting in diminished production of the TCA cycle intermediates oxaloacetate and NADPH, and impaired oxygen consumption. The defective GSIS and mitochondrial metabolism in MDM2-null islets can be rescued by restoring PC expression. Under diabetogenic conditions, MDM2 and p53 are upregulated, whereas PC is reduced in mouse b-cells. Pharmacological inhibition of p53 alleviates defective GSIS in diabetic islets by restoring PC expression. Thus, the MDM2–p53–PC signalling axis links mitochondrial metabolism to insulin secretion and glucose homeostasis, and could represent a therapeutic target in diabetes. 1 State Key Laboratory of Pharmaceutical Biotechnology, The University of Hong Kong, Pok Fu Lam, Hong Kong. 2 Department of Medicine, The University of Hong Kong, Pok Fu Lam, Hong Kong. -

Pyruvate Carboxylase and Phosphoenolpyruvate Carboxykinase Activity in Leukocytes and Fibroblasts from a Patient with Pyruvate Carboxylase Deficiency
Pediat. Res. 13: 3843 (1979) Citrate synthase lymphocytes fibroblasts phosphoenol pyruvate carboxykinase lactic acidosis pyruvate carboxylase pyruvate carboxylase deficiency Pyruvate Carboxylase and Phosphoenolpyruvate Carboxykinase Activity in Leukocytes and Fibroblasts from a Patient with Pyruvate Carboxylase Deficiency Division of Genetics and Departmenr of Pediatrics, Universiy of Oregon Health Sciences Center, Porrland, Oregon; and Department of Biochemistry, School of Medicine, Case Western Reserve Universi!,., Cleveland, Ohio, USA Summary complex (PDC) (7, 13, 32, 35), and PEPCK (14, 17,40). With the exception of PDC which catalyzes the first step in the Krebs cycle, Normal values are given for the activities of pyruvate carbox- the enzymes are necessary for gluconeogenesis in mammalian ylase (E.C. 6.4.1.1), mitochondrial phosphoenolpyruvate carboxy- renal cortex and liver. In addition to its gluconeogenic role. base (E.C. 4.1.1.32, PEPCK), and citrate synthase (E.C. 4.1.3.7) pyruvate carboxylase is required for the adequate operation of the in fibroblasts, lymphocytes, and leukocytes. Also given are values Krebs cycle because of its role in the replenishment of four-carbon for these enzymes in the leukocytes and fibroblasts from a severely Krebs cycle intermediates. Both the kitochondrial and cytosolic mentally and developmentally retarded patient with proximal renal forms of PEPCK are present in human liver. tubular acidosis and hepatic, cerebral, and renal cortical pyruvate Fructose 1,6-bisphosphatase (28) and PDC deficiency (7) have carboxylase deficiency. In normals, virtually all of the mitochon- been diagnosed in leukocytes and, although there are references drial PEPCK and pyruvate carboxylase activity was present in the to the presence of pyruvate carboxylase in fibroblasts (3. -

Nuclear Magnetic Resonance Approaches in the Study of 2-Oxo Acid Dehydrogenase Multienzyme Complexes— a Literature Review
Molecules 2013, 18, 11873-11903; doi:10.3390/molecules181011873 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Nuclear Magnetic Resonance Approaches in the Study of 2-Oxo Acid Dehydrogenase Multienzyme Complexes— A Literature Review Sowmini Kumaran 1, Mulchand S. Patel 2 and Frank Jordan 1,* 1 Department of Chemistry, Rutgers University, Newark, NJ 07102, USA 2 Department of Biochemistry, School of Medicine and Biomedical Sciences, State University of New York at Buffalo, Buffalo, NY 14214, USA * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-973-353-5470; Fax: +1-973-353-1264. Received: 30 July 2013; in revised form: 14 September 2013 / Accepted: 16 September 2013 / Published: 26 September 2013 Abstract: The 2-oxoacid dehydrogenase complexes (ODHc) consist of multiple copies of three enzyme components: E1, a 2-oxoacid decarboxylase; E2, dihydrolipoyl acyl-transferase; and E3, dihydrolipoyl dehydrogenase, that together catalyze the oxidative decarboxylation of 2-oxoacids, in the presence of thiamin diphosphate (ThDP), coenzyme A 2+ + (CoA), Mg and NAD , to generate CO2, NADH and the corresponding acyl-CoA. The structural scaffold of the complex is provided by E2, with E1 and E3 bound around the periphery. The three principal members of the family are pyruvate dehydrogenase (PDHc), 2-oxoglutarate dehydrogenase (OGDHc) and branched-chain 2-oxo acid dehydrogenase (BCKDHc). In this review, we report application of NMR-based approaches to both mechanistic and structural issues concerning these complexes. These studies revealed the nature and reactivity of transient intermediates on the enzymatic pathway and provided site-specific information on the architecture and binding specificity of the domain interfaces using solubilized truncated domain constructs of the multi-domain E2 component in its interactions with the E1 and E3 components. -

Recent Progress in the Microbial Production of Pyruvic Acid
fermentation Review Recent Progress in the Microbial Production of Pyruvic Acid Neda Maleki 1 and Mark A. Eiteman 2,* 1 Department of Food Science, Engineering and Technology, University of Tehran, Karaj 31587-77871, Iran; [email protected] 2 School of Chemical, Materials and Biomedical Engineering, University of Georgia, Athens, GA 30602, USA * Correspondence: [email protected]; Tel.: +1-706-542-0833 Academic Editor: Gunnar Lidén Received: 10 January 2017; Accepted: 6 February 2017; Published: 13 February 2017 Abstract: Pyruvic acid (pyruvate) is a cellular metabolite found at the biochemical junction of glycolysis and the tricarboxylic acid cycle. Pyruvate is used in food, cosmetics, pharmaceutical and agricultural applications. Microbial production of pyruvate from either yeast or bacteria relies on restricting the natural catabolism of pyruvate, while also limiting the accumulation of the numerous potential by-products. In this review we describe research to improve pyruvate formation which has targeted both strain development and process development. Strain development requires an understanding of carbohydrate metabolism and the many competing enzymes which use pyruvate as a substrate, and it often combines classical mutation/isolation approaches with modern metabolic engineering strategies. Process development requires an understanding of operational modes and their differing effects on microbial growth and product formation. Keywords: auxotrophy; Candida glabrata; Escherichia coli; fed-batch; metabolic engineering; pyruvate; pyruvate dehydrogenase 1. Introduction Pyruvic acid (pyruvate at neutral pH) is a three carbon oxo-monocarboxylic acid, also known as 2-oxopropanoic acid, 2-ketopropionic acid or acetylformic acid. Pyruvate is biochemically located at the end of glycolysis and entry into the tricarboxylic acid (TCA) cycle (Figure1). -

(12) United States Patent (10) Patent No.: US 7927,859 B2 San Et Al
USOO7927859B2 (12) United States Patent (10) Patent No.: US 7927,859 B2 San et al. (45) Date of Patent: Apr. 19, 2011 (54) HIGH MOLARSUCCINATEYIELD FOREIGN PATENT DOCUMENTS BACTERIA BY INCREASING THE WO WO99.06532 * 2/1999 INTRACELLULAR NADHAVAILABILITY WO WO 2007 OO1982 1, 2007 OTHER PUBLICATIONS (75) Inventors: Ka-Yiu San, Houston, TX (US); George N. Bennett, Houston, TX (US); Ailen Branden et al. Introduction to Protein Structure, Garland Publishing Inc., New York, p. 247, 1991.* Sánchez, Houston, TX (US) ExPASy. Formate Dehydrogenase.* Vemuri et al. Effects of growth mode and pyruvate carboxylase on (73) Assignee: Rice University, Houston, TX (US) Succinic acid production by metabolically engineered strains of Escherichia coli. Appl Environ Microbiol. Apr. 2002:68(4): 1715 (*) Notice: Subject to any disclaimer, the term of this 27.3 patent is extended or adjusted under 35 Goodbye et al. Cloning and sequence analysis of the fermentative alcohol-dehydrogenase-encoding gene of Escherichia coli. Gene. U.S.C. 154(b) by 791 days. Dec. 21, 1989;85(1):209-14.* Datsenko et al. One-step inactivation of chromosomal genes in (21) Appl. No.: 10/923,635 Escherichia coli K-12 using PCR products. Proc Natl AcadSci USA. Jun. 6, 2000:97(12):6640-5.* (22) Filed: Aug. 20, 2004 Berrios-Rivera et al. Metabolic engineering of Escherichia coli: increase of NADHavailability by overexpressing an NAD(+)-depen (65) Prior Publication Data dent formate dehydrogenase. Metab Eng. Jul. 2002;4(3):217-29.* Gupta et al. Escherichia coli derivatives lacking both alcohol US 2005/OO42736A1 Feb. 24, 2005 dehydrogenase and phosphotransacetylase grow anaerobically by lactate fermentation. -

Assessing the Thiamine Diphosphate Dependent Pyruvate Dehydrogenase E1 Subunit for Carboligation Reactions with Aliphatic Ketoacids
International Journal of Molecular Sciences Article Assessing the Thiamine Diphosphate Dependent Pyruvate Dehydrogenase E1 Subunit for Carboligation Reactions with Aliphatic Ketoacids Stefan R. Marsden, Duncan G. G. McMillan and Ulf Hanefeld * Biokatalyse, Afdeling Biotechnologie, Technische Universiteit Delft, Van der Maasweg 9, 2629HZ Delft, The Netherlands; [email protected] (S.R.M.); [email protected] (D.G.G.M.) * Correspondence: [email protected] Received: 12 October 2020; Accepted: 12 November 2020; Published: 16 November 2020 Abstract: The synthetic properties of the Thiamine diphosphate (ThDP)-dependent pyruvate dehydrogenase E1 subunit from Escherichia coli (EcPDH E1) was assessed for carboligation reactions with aliphatic ketoacids. Due to its role in metabolism, EcPDH E1 was previously characterised with respect to its biochemical properties, but it was never applied for synthetic purposes. Here, we show that EcPDH E1 is a promising biocatalyst for the production of chiral α-hydroxyketones. WT EcPDH E1 shows a 180–250-fold higher catalytic efficiency towards 2-oxobutyrate or pyruvate, respectively, in comparison to engineered transketolase variants from Geobacillus stearothermophilus (TKGST). Its broad active site cleft allows for the efficient conversion of both (R)- and (S)-configured α-hydroxyaldehydes, next to linear and branched aliphatic aldehydes as acceptor substrates under kinetically controlled conditions. The alternate, thermodynamically controlled self-reaction of aliphatic aldehydes was shown to be limited to low levels of conversion, which we propose to be due to their large hydration constants. Additionally, the thermodynamically controlled approach was demonstrated to suffer from a loss of stereoselectivity, which makes it unfeasible for aliphatic substrates. Keywords: Thiamine diphosphate; transketolase; C-C bond formation; kinetic control; acyloins 1.