A Functional Landscape of CKD Entities from Public Transcriptomic Data
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Validation of a Prospective Urinalysis-Based Prediction Model for ICU Resources and Outcome of COVID-19 Disease: a Multicenter Cohort Study
Journal of Clinical Medicine Article Validation of a Prospective Urinalysis-Based Prediction Model for ICU Resources and Outcome of COVID-19 Disease: A Multicenter Cohort Study Oliver Gross 1,* , Onnen Moerer 2,† , Thomas Rauen 3,†, Jan Böckhaus 1,†, Elion Hoxha 4,†, Achim Jörres 5, Matthias Kamm 5, Amin Elfanish 5, Wolfram Windisch 6, Michael Dreher 7,‡, Juergen Floege 3, Stefan Kluge 8, Christian Schmidt-Lauber 4, Jan-Eric Turner 4, Samuel Huber 9 , Marylyn M. Addo 9 , Simone Scheithauer 10, Tim Friede 11,§ , Gerald S. Braun 3,§, Tobias B. Huber 4,§ and Sabine Blaschke 12,§ 1 Clinic for Nephrology and Rheumatology, University Medical Center Göttingen, 37075 Göttingen, Germany; [email protected] 2 Clinic of Anaesthesiology, University Medical Center Göttingen, 37075 Göttingen, Germany; [email protected] 3 Division of Nephrology and Clinical Immunology, RWTH Aachen University, 52074 Aachen, Germany; [email protected] (T.R.); jfl[email protected] (J.F.); [email protected] (G.S.B.) 4 III. Department of Medicine, University Medical Center Hamburg-Eppendorf, 20246 Hamburg, Germany; [email protected] (E.H.); [email protected] (C.S.-L.); [email protected] (J.-E.T.); [email protected] (T.B.H.) 5 Department of Medicine I, Nephrology, Transplantation and Medical Intensive Care, Citation: Gross, O.; Moerer, O.; University Witten/Herdecke Medical Center Cologne-Merheim, 51109 Cologne, Germany; Rauen, T.; Böckhaus, J.; Hoxha, E.; [email protected] (A.J.); [email protected] (M.K.); [email protected] (A.E.) 6 Jörres, A.; Kamm, M.; Elfanish, A.; Department of Pneumology and Critical Care Medicine, University Witten/Herdecke, Medical Center Cologne-Merheim, 51109 Cologne, Germany; [email protected] Windisch, W.; Dreher, M.; et al. -

Dr. Martin Zimmermann Curriculum Vitae
Dr. Martin Zimmermann Curriculum Vitae Professional Mail: Contact Reactive Systems Group Universität des Saarlandes 66123 Saarbrücken Germany Office: Saarland Informatics Campus, Building E 1 1, Room 1.13 Phone: +49 681 302 5667 Email: [email protected] URL: www.react.uni-saarland.de/people/zimmermann.html Employment Saarland University May 2013 - present Postdoc University of Warsaw February 2012 - April 2013 Postdoc RWTH Aachen University February 2009 - January 2012 Research Assistant Education RWTH Aachen University February 2009 - January 2012 PhD Student Thesis: Solving Infinite Games with Bounds Adviser: Wolfgang Thomas RWTH Aachen University September 2003 - January 2009 Diploma in Computer Science Minor in Business Administration Thesis: Time-optimal Winning Strategies in Infinite Games Adviser: Wolfgang Thomas Awards and Springorum Medal 2010 Scholarships Awarded for diploma with distinction at RWTH Aachen University Fulbright Scholarship September 2007 - June 2008 DePaul University, Chicago, IL GPA (through one year): 4.0 Grants DFG Project “Tradeoffs in Controller Synthesis” January 2015 - September 2018 Principal Investigator Grant covers salary and travel for PI and one PhD student 1 Activities GandALF 2018 PC co-chair and organizing chair Highlights of Logic, Games, and Automata 2018 PC member TIME 2017 PC member Events Workshop “Algorithmic Verification of Real-time Systems” December 2016 Invited Speaker Workshop “Automata, Concurrency and Timed Systems” February 2015 Invited Speaker Dagstuhl Seminar “Non-Zero-Sum-Games -

Excellence in Engineering and Science Made in Germany TU9 Alliance
Excellence in Engineering and Science Made in Germany TU9 Alliance Technische Leibniz Universität University Berlin Hannover Technische Universität Braunschweig Technische RWTH Universität Aachen Dresden University Technische Universität Darmstadt Karlsruhe Institute of Technology Technical University University of Stuttgart of Munich TU9 Mission Statement TU9 — German Universities of Technology Excellence in Engineering and Science Made in Germany TU9 is the Alliance of leading Universities of Technology in Germany: RWTH Aachen University, Technische Universität Berlin, Technische Universität Braunschweig, Technische Universität Darmstadt, Technische Universität Dresden, Leibniz University Hannover, Karlsruhe Institute of Technology, Technical University of Munich, and University of Stuttgart. Tradition, excellence, and innovation are the hallmarks of TU9 Universities. Founded during the Industrial Age, they contributed decisively to tech nological progress back then and continue to do so today. They enjoy an outstanding reputation around the world as renowned research and teaching institutions that promote the transfer of knowledge and tech nology between universities and practice. As such, they train exceptional young academics for careers in science, business, and administration and assume social responsibility. TU9 Universities foster top-class inter- national networks and diverse cooperation with industry, making them a key element of the German science and innovation landscape. The excellent research and teaching at TU9 Universities are based on independence, plurality, and freedom of expression. TU9 Universities have always been places of intellectual and cultural diversity where inter nationalization and integration are a matter of course. TU9 Universities embody ▪ tradition & innovation, ▪ excellence & interdisciplinarity, ▪ cooperation & competencies, and the world of tomorrow. TU9 Universities … excel in pioneering creative research in engineering and the natural sciences. -

Janet's Algorithm and Systems Theory
Janet’s Algorithm and Systems Theory Daniel Robertz Lehrstuhl B f¨ur Mathematik http://wwwb.math.rwth-aachen.de/~daniel 28.11.2012 Inria Saclay – ˆIle-de-France / L2S, Sup´elec, 28.11.2012 Outline Janet’s algorithm Module-theoretic approach to linear systems Parametrizing linear systems Inria Saclay – ˆIle-de-France / L2S, Sup´elec, 28.11.2012 Janet’s algorithm Inria Saclay – ˆIle-de-France / L2S, Sup´elec, 28.11.2012 Janet’s algorithm for linear PDEs ∂2u ∂u − = 0 ∂x∂y ∂y find: u = u(x,y) analytic ∂2u ∂u − = 0 ∂x2 ∂y 2 x2 xy y u(x,y)= a0,0 + a1,0 x + a0,1 y + a2,0 2! + a1,1 1!1! + a0,2 2! + ... Inria Saclay – ˆIle-de-France / L2S, Sup´elec, 28.11.2012 Janet’s algorithm for linear PDEs ∂2u ∂u − = 0 ∂x∂y ∂y find: u = u(x,y) analytic ∂2u ∂u − = 0 ∂x2 ∂y 2 x2 xy y u(x,y)= a0,0 + a1,0 x + a0,1 y + a2,0 2! + a1,1 1!1! + a0,2 2! + ... Inria Saclay – ˆIle-de-France / L2S, Sup´elec, 28.11.2012 Janet’s algorithm for linear PDEs ∂2u ∂u − = 0 ∂x∂y ∂y find: u = u(x,y) analytic ∂2u ∂u − = 0 ∂x2 ∂y ∂ ∂2u ∂u ∂ ∂2u ∂u ∂2u ∂u ∂x ∂x∂y − ∂y − ∂y ∂x2 − ∂y = ∂y2 − ∂y = 0 2 x2 xy y u(x,y)= a0,0 + a1,0 x + a0,1 y + a2,0 2! + a1,1 1!1! + a0,2 2! + ... Inria Saclay – ˆIle-de-France / L2S, Sup´elec, 28.11.2012 Janet’s algorithm for linear PDEs ∂2u ∂u − = 0 ∂x∂y ∂y find: u = u(x,y) analytic ∂2u ∂u − = 0 ∂x2 ∂y ∂ ∂2u ∂u ∂ ∂2u ∂u ∂2u ∂u ∂x ∂x∂y − ∂y − ∂y ∂x2 − ∂y = ∂y2 − ∂y = 0 2 x2 xy y u(x,y)= a0,0 + a1,0 x + a0,1 y + a2,0 2! + a1,1 1!1! + a0,2 2! + .. -

RWTH Aachen University Engineering Summer Schools 2017
RWTH Aachen University Engineering Summer Schools 2017 Studying at one of the best German Universities in Engineering! Welcome Summer Programmes Why choose us The City of Aachen Welcome to RWTH International Academy A short summary of what we have to offer Welcome All Summer School courses offered by RWTH International Academy are uniquely designed for international undergraduate students who want to gain insight into one of Germany’s Universities of Excellence. The RWTH International Academy works closely together with various Institutes of the RWTH Aachen University, the International Office and the Language Center to offer international students a perfect mix of technical and practical knowledge as well as cultural experiences. Excellent Science and Our Summer School courses in Mechanical Engineering and Management offer international Research students the opportunity to take part in excellent science and research at the RWTH Aachen University. The University is highly acclaimed internationally for its development of innovative answers to the most pressing global challenges. Its success in Germany‘s Excellence Initiative Experience Excellence in Science and Engineering at the RWTH Aachen University reflects its outstanding reputation for research and education in the fields of science, technology, RWTH Aachen University is the largest university of technology in Germany and the most important economic factor of the city. and engineering. Every year, a large number of international students and scientists come to the university to benefit from its high-quality study programmes German Language and Culture Through the university’s “BeBuddy” programme, international students have the chance to im- and excellent facilities, from which both are internationally recognised. -

Curriculum Vitae
Curriculum Vitae Prof. Dr. rer. nat. Egbert Figgemeier Birth date: 04.02.1971 RWTH Aachen University Helmholtz-Institut Münster – IEK-12 Institute for Power Electronics & Electrical Drives (ISEA) Professor for Ageing and Lifetime Prediction of Batteries Jaegerstrasse 17-19, 52066 Aachen / Germany Phone +49 241 80-97158 Fax +49 241 80-92203 eMail [email protected] Work Experience – Academic & Industry Since W2 Professorship at RWTH Aachen University (Faculty of Electric Engineering) 05/2016 and group leader Helmholtz-Institute Münster (Research Center Juelich) “Aging and Reliability of Batteries” 2012 - 2016 Senior application engineer for battery and energy storage materials at 3M Germany, Neuss 2007 – Lab Head and Manager at Bayer Technology Services, GmbH, Leverkusen, 2012 Germany 2004 – Group leader and Treubel Foundation Fellow at the University of Basel, 2007 Switzerland 2001 – Researcher and Marie-Curie-Fellow at the University of Uppsala, Sweden 2004 1998 – Post-Doc at Dublin City University, Dublin, Ireland and at University of Basel, 2000 Switzerland 1998 PhD in Physical Chemistry at University of Paderborn 1996 Diploma in Chemistry at University of Paderborn Selected Key Activities Investigation of degradation mechanisms in Lithium ion battery cells Materials and product development at 3M and Bayer Key expert at 3M and Bayer for energy storage materials Business development manager for energy storage materials at 3M and Bayer Material Science R&D work at the Universities of Dublin, Basel and Uppsala in -

Simon, Ulrich, Prof. Dr. Rer. Nat
Simon, Ulrich, Prof. Dr. rer. nat. * 18 October 1963, male Institut für Anorganische Chemie RWTH Aachen University, 52074 Aachen Phone: +49 (0) 241 80 94644 E-mail: [email protected] Current position: Professor (W3) University training and degree 1983–1990 Degree in chemistry (summa cum laude), thesis in solid state chemistry, Universität Duisburg-Essen Advanced academic qualifications 1992 Doctorate in inorganic chemistry (summa cum laude), Universität Duisburg-Essen Postgraduate professional career 2018–2020 Dean of the Faculty of Mathematics, Computer Science and Natural Sciences 2012–2016 Chairman of the Senate at RWTH Aachen University 2012 Visiting professor, International Institute for Nanotechnology, Northwestern University, Evanston, USA (1 month) Since 2011 Steering committee member of the DFG collaborative research center Nanoswitches (SFB 917) 2008–2010 Dean of the Faculty of Mathematics, Computer Science and Natural Sciences 2007 Visiting professor, International Institute for Nanotechnology, Northwestern University, Evanston, USA (3 months) 2006–2008 Vice-dean of the Faculty of Mathematics, Computer Science and Natural Sciences 2004–2006 Head of the Department of Chemistry, RWTH Aachen University since 2000 Chair of Inorganic Chemistry and Electrochemistry, RWTH Aachen University 1999–2000 Associate professor, Universität Duisburg-Essen 1992–1999 Assistant professor, Universität Duisburg-Essen Other • Member of the Cluster of Excellence “The Fuel Science Center” (since 2018) • RWTH Fellow award for experienced -

Robotic Leg Illusion: System Design and Human-In-The-Loop Evaluation Dimitri Penner, Anna M
372 IEEE TRANSACTIONS ON HUMAN-MACHINE SYSTEMS, VOL. 49, NO. 4, AUGUST 2019 Robotic Leg Illusion: System Design and Human-in-the-Loop Evaluation Dimitri Penner, Anna M. H. Abrams, Philipp Overath , Joachim Vogt, and Philipp Beckerle , Member, IEEE Abstract—The question how humans integrate artificial lower mobility. Besides physical challenges of getting used to the limb devices into their body schema has distinct potential for engi- prosthesis, users have to psychologically and socially adapt to neering motion assistance systems, e.g., the design of robotic pros- a new self-image [2]. A prosthesis that resembles appearance theses. Adding robotic technology to existing psychological exper- iments enables a deeper investigation of multisensory interaction and function of the real extremity might help to counteract neg- between proprioceptive, visual, and tactile stimuli during motion. ative psychological consequences [3]. Robotic technology is a This paper reports the design and control of a robot to investigate very promising means to improve prosthetic function [4]. Nev- embodiment with regard to the lower limbs. In an evaluation study, ertheless, all users undergo two central psychological changes the rubber hand illusion is transferred to the whole leg for the first concerning their identity: changes in perception of the own body time. Participants performed knee bends according to three differ- ent conditions being imitated by a robotic leg. The occurrence of and integration of the prosthesis into their body schema [5]. a robotic leg illusion was subjectively assessed by a questionnaire Body perception is influenced by physical, sensory, and social and objectively measured by the proprioceptive drift. Consider- factors. -

Call-For-Papers Lim2021 NEW.Pdf
Scientific Committee General Chair J. P. Bergmann, Technische Universität Ilmenau, Germany Michael Rethmeier M. Brandt, RMIT University, Australia Bundesanstalt für Materialforschung und -prüfung (BAM) Berlin, Germany D. Drummer, Universität Erlangen-Nürnberg, Germany C. Emmelmann, Hamburg University of Technology, Germany Conference Office C. Esen, Ruhr-Universität Bochum, Germany A. Gillner, RWTH Aachen University, Germany Hans-Joachim Krauß | Stephanie Wiedenmann Bayerisches Laserzentrum GmbH , University of Stuttgart, Germany T. Graf Erlangen, Germany B. Huis in 't Veld, Fontys Univ. of Applied Sci., The Netherlands S. Kaierle, Laser Zentrum Hannover e.V., Germany Tel.: +49 9131 97790 -23 (-0) E-mail: [email protected] , Luleå University of Technology, Sweden A. Kaplan URL: www.wlt.de/lim R. Kling, ALPhANOV, France P. Krakhmalev, Karlstad University, Sweden Conference Venue H.-J. Krauß, Bayerisches Laserzentrum GmbH, Germany ICM – Internationales Congress Center München A.-F. Lasagni, Technische Universität Dresden, Germany Messegelände C. Leyens, Fraunhofer IWS, Germany 81823 Munich S.-J. Na, Xi'an Jiaotong University, China Germany B. Neuenschwander, Bern Univ. of Applied Sci., Switzerland URL: www.icm-muenchen.de S. Nolte, Friedrich Schiller University Jena, Germany Y. Okamoto, Okayama University, Japan Conference Fees A. Olowinsky, Fraunhofer ILT, Germany Included are the conference proceedings, the LiM social event, A. Ostendorf, Ruhr-Universität Bochum, Germany visit of the trade fair, and participation at all World of Photonics A. Otto, Vienna University of Technology, Austria Congress conferences. L. Overmeyer, Laser Zentrum Hannover e.V., Germany We kindly ask for your understanding that the calculation of the J. Pou, University of Vigo, Spain conference fees is currently a dynamic process, depending on U. -

RWTH Aachen University an Excellent Choice for Your Stay Abroad!
Foto: Martin Braun RWTH Aachen University An Excellent Choice for Your Stay Abroad! Talk by Alexander Mallmann Why should one study at RWTH Aachen University? What makes RWTH Aachen University special ? • One of eleven exelence Universities of Germnay • 5 Noble prize winner • 46k> Students (over 6000 new Bachelor Students/year) + 10k> employees • 150 years of Tradition and expertise • Scientfic and industrial integration → Franuhofer and Leibniz insitutes → Research centers( e.g. Jülich) → Collabrations with other universities (e.g. ETH, Aalto, Delft, University of Tokyo) → Business Partners (e.g. E-Go , BMW, Lufthansa) 3 RWTH Aachen University – An Excellent Choice for your Stay Abroad! | 2021 Facts about RWTH Aachen University Inexpensive German 25 % International Students Language Courses 46k Students in total No Tuition Fee 157+ Study Programs Lots of Courses taught in English 4 RWTH Aachen University – An Excellent Choice for your Stay Abroad! | 2021 Services for Enrolled Students Semester Fee (~ 290 €) including: Semester Ticket Accident Insurance Student Discounts • Sport Activities • Cinemas, Theatre • Museums, etc. 5 RWTH Aachen University – An Excellent Choice for your Stay Abroad! | 2021 RWTH Semester Ticket Free Public Transportation in NRW! Düsseldorf Köln Aachen © Wikipedia © https://www.duesseldorf.de/ 6 RWTH Aachen University – An Excellent Choice for your Stay Abroad! | 2021 Comparison Aalto University vs RWTH Aachen University • Campus University with Teekkarikylä • Delocalized Campus, University Buildings are spread -
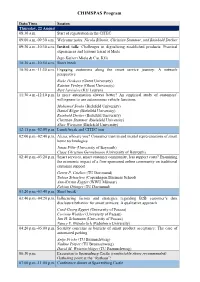
CHIMSPAS Program
CHIMSPAS Program Date/Time Session Thursday, 22 August 08:30 a.m. Start of registration in the CITEC 09:00 a.m.-09:30 a.m. Welcome notes: Nicola Bilstein, Christian Stummer, and Reinhold Decker 09:30 a.m.-10:30 a.m. Invited talk: Challenges in digitalizing established products: Practical experiences and lessons learnt at Miele Ingo Kaiser (Miele & Cie. KG) 10:30 a.m.-10:50 a.m. Short break 10:50 a.m.-11:30 a.m. Engaging customers along the smart service journey: A network perspective Bieke Henkens (Ghent University) Katrien Verleye (Ghent University) Bart Larivière (KU Leuven) 11:30 a.m.-12:10 p.m. Is more automation always better? An empirical study of customers’ willingness to use autonomous vehicle functions Mohamed Souka (Bielefeld University) Daniel Böger (Bielefeld University) Reinhold Decker (Bielefeld University) Christian Stummer (Bielefeld University) Alisa Wiemann (Bielefeld University) 12:15 p.m.-02:00 p.m. Lunch break and CITEC tour 02:00 p.m.-02:40 p.m. Alexa, who are you? Consumer trust in and mental representations of smart home technologies Jonas Föhr (University of Bayreuth) Claas Christian Germelmann (University of Bayreuth) 02:40 p.m.-03:20 p.m. Smart services, smart customer community, less support costs? Examining the economic impact of a firm-sponsored online community on traditional customer support Gerrit P. Cziehso (TU Dortmund) Tobias Schaefers (Copenhagen Business School) Ann-Kristin Kupfer (WWU Münster) Fabian Ottinger (TU Dortmund) 03:20 p.m.-03:40 p.m. Short break 03:40 p.m.-04:20 p.m. Influencing factors and strategies regarding B2B customer’s data disclosure behavior for smart services: A qualitative approach Curd-Georg Eggert (University of Passau) Corinna Winkler (University of Passau) Jan H. -

CV Dr. (Des) Pascale Willemsen
CV Dr. (des) Pascale Willemsen Current Position Postdoctoral researcher Ruhr-University Bochum Institute for Philosophy II, Chair for Philosophy of Mind Center for Mind, Brain, and Cognitive Evolution Universitaetsst. 150 44801 Bochum PascaleWillemsen.com Areas of Specialization Philosophy of Moral Psychology, Philosophy of Mind, Experimental Philosophy Areas of Competence Ethics, Philosophy of Action Previous Academic Positions 10/2017 – 03/2018 Postdoctoral researcher University College London Department for Experimental Psychology 06/2017 – 09/2017 Postdoctoral researcher Ruhr-University Bochum Institute for Philosophy II, Chair for Philosophy of Mind Education 01/2014 – 06/2017 PhD in Philosophy Ruhr-University Bochum Thesis: “Investigating the Role of Causal Responsibility for the Attribution of Moral Responsibility” Committee: Prof. Albert Newen (first supervisor), Prof. Edouard Machery (second supervisor), Prof. Corinna Mieth, Prof. Tobias Schlicht, Prof. James Wilberding Grade: summa cum laude (highest possible distinction) 09/2015 – 11/2015 Visiting Fellow, Yale University Host: Prof. Joshua Knobe 09/2013 MSc. in Philosophy of Science London School of Economics and Political Sciences, London 09/2012 M.A. in Philosophy M.A. in Linguistics and Communication Studies RWTH Aachen University, Aachen 08/2010 B.A. in Philosophy B.A. in Linguistics and Communication Studies RWTH Aachen University, Aachen Books In press Willemsen, Pascale. “Investigating the Role of Causal Responsibility for the Attribution of Moral Responsibility”, accepted for publication with mentis. Peer-Reviewed Research Articles 2018 Willemsen, Pascale. “The Relevance of Alternative Possibilities for the Attribution of Moral Responsibility”, submitted to Oxford Series in Experimental Philosophy, Oxford University Press. Revision in progress. Willemsen, Pascale; Kirfel, Lara. “Recent empirical work on causation”, submitted to Philosophy Compass.