(BMP) 4 Signaling Antagonist in Controlling Mouse Lung Development
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Redalyc.Possible Role of Noggin Gene in Mandibular Development
Universitas Odontológica ISSN: 0120-4319 [email protected] Pontificia Universidad Javeriana Colombia Gutiérrez Prieto, Sandra; Torres López, Diana; Gómez Rodríguez, Mariluz; García Robayo, Adriana Possible Role of Noggin Gene in Mandibular Development Universitas Odontológica, vol. 34, núm. 73, julio-diciembre, 2015, pp. 21-31 Pontificia Universidad Javeriana Bogotá, Colombia Available in: http://www.redalyc.org/articulo.oa?id=231247071014 How to cite Complete issue Scientific Information System More information about this article Network of Scientific Journals from Latin America, the Caribbean, Spain and Portugal Journal's homepage in redalyc.org Non-profit academic project, developed under the open access initiative Possible Role of Noggin Gene in Mandibular Development Posible papel del gen noggin en el desarrollo mandibular Possível papel do gene noggin no desenvolvimento mandibular 21 Sandra Gutiérrez Prieto ABSTRACT Odontóloga, magistra y PhD en Background: Noggin (Nog) gene is one of the antagonists of bone morphogenic proteins (BMPs) Ciencias Biológicas, docente and its function is to modulate the signs. When Nog’s action is ineffective, an excessive activity investigadora del Centro de of BMPs occur causing serious developmental abnormalities. Studies have shown that Nog is Investigaciones Odontológicas, critical for chondrogenesis, osteogenesis, and joint training and appears to be involved in the 2027-3444 | e-ISSN 0120-4319 ISSN . Pontificia Universidad Javeriana, growth of craniofacial structures, including the jaw. There are in the literature a few studies Bogotá, Colombia. about the relationship between Nog and its role in mandibular development. Purpose: To r e - views the molecular factors involved in the jaw development. Method: Focusing primarily on Diana Torres López BMPs, their function, and signaling pathway as Nog regulates this path. -

In Vivo Imaging of Tgfβ Signalling Components Using Positron
REVIEWS Drug Discovery Today Volume 24, Number 12 December 2019 Reviews KEYNOTE REVIEW In vivo imaging of TGFb signalling components using positron emission tomography 1 1 2 Lonneke Rotteveel Lonneke Rotteveel , Alex J. Poot , Harm Jan Bogaard , received her MSc in drug 3 1 discovery and safety at the Peter ten Dijke , Adriaan A. Lammertsma and VU University in 1 Amsterdam. She is Albert D. Windhorst currently finishing her PhD at the VU University 1 Department of Radiology and Nuclear Medicine, Amsterdam UMC, location VUmc, Amsterdam, The Netherlands Medical Center (VUmc) 2 under the supervision of A. Pulmonary Medicine, Institute for Cardiovascular Research, Amsterdam UMC, location VUmc, Amsterdam, The Netherlands D. Windhorst and Adriaan A. Lammertsma. Her 3 research interest is on the development of positron Department of Cell and Chemical Biology, Oncode Institute, Leiden University Medical Center, Leiden, The emission tomography (PET) tracers that target Netherlands selectively the activin receptor-like kinase 5 in vitro and in vivo. Alex J. Poot obtained his The transforming growth factor b (TGFb) family of cytokines achieves PhD in medicinal chemistry homeostasis through a careful balance and crosstalk with complex from Utrecht University. As postdoctoral researcher at signalling pathways. Inappropriate activation or inhibition of this pathway the VUmc, Amsterdam, he and mutations in its components are related to diseases such as cancer, developed radiolabelled anticancer drugs for PET vascular diseases, and developmental disorders. Quantitative imaging of imaging. In 2014, he accepted a research expression levels of key regulators within this pathway using positron fellowship from Memorial Sloan Kettering Cancer 13 emission tomography (PET) can provide insights into the role of this Center, New York to develop C-labelled probes for tumour metabolism imaging with magnetic resonance in vivo pathway , providing information on underlying pathophysiological imaging (MRI). -

Role of TGF-Β3 in the Regulation of Immune Responses T
Role of TGF-β3 in the regulation of immune responses T. Okamura1,2, K. Morita1, Y. Iwasaki1, M. Inoue1, T. Komai1, K. Fujio1, K. Yamamoto1 1Department of Allergy and ABSTRACT ences, indicating their non-redundancy Rheumatology, Graduate School of Transforming growth factor-betas (12-16). The TGF-βs have opposite ef- Medicine, The University of Tokyo, (TGF-βs) are multifunctional cytokines fects on tissue fibrosis. Wound-healing Tokyo, Japan; that have been implicated in the regu- experiments revealed that TGF-β1 2Max Planck-The University of Tokyo Center for Integrative Inflammology, lation of a broad range of biological and TGF-β2 cause fibrotic scarring The University of Tokyo, Tokyo, Japan. processes, including cell proliferation, responses and that TGF-β3 induces a Tomohisa Okamura, MD, PhD cell survival, and cell differentiation. scar-free response (17). Both TGF-β1 Kaoru Morita, MD The three isoforms identified in mam- and -β2 activate the collagen α2 (I) Yukiko Iwasaki, MD, PhD mals, TGF-β1, TGF-β2, and TGF-β3, gene promoter, resulting in increased Mariko Inoue, MD have high sequence homology, bind to collagen synthesis (18). It was also re- Toshihiko Komai, MD the same receptors, and show similar ported that TGF-β1, but not TGF-β3, is Keishi Fujio, MD, PhD biological functions in many in vitro a crucial factor in the development of Kazuhiko Yamamoto, MD, PhD studies. However, analysis of the in vivo pulmonary fibrosis (19). Most of the in- Please address correspondence to: functions of the three isoforms and mice formation on the immunological activ- Keishi Fujio, MD, PhD, deficient for each cytokine reveals strik- Department of Allergy and ity of TGF-βs derives from studies of Rheumatology, ing differences, illustrating their unique TGF-β1 and, in part, TGF-β2, whereas Graduate School of Medicine, biological importance and functional recent investigations have begun to il- The University of Tokyo, non-redundancy. -

The Role of the TGF- Co-Receptor Endoglin in Cancer
1 The role of the TGF- co-receptor endoglin in cancer Eduardo Pérez-Gómez1,†, Gaelle del Castillo1, Juan Francisco Santibáñez2, Jose Miguel López-Novoa3, Carmelo Bernabéu4 and 1,* Miguel Quintanilla . 1Instituto de Investigaciones Biomédicas Alberto Sols, Consejo Superior de Investigaciones Científicas (CSIC)-Universidad Autónoma de Madrid, 28029-Madrid, Spain; 2Institute for Medical Research, University of Belgrado, Belgrado, Serbia; 3Instituto Reina Sofía de Investigación Nefrológica, Departamento de Fisiología y Farmacología, Universidad de Salamanca, Salamanca, Spain; 4Centro de Investigaciones Biológicas, CSIC, and CIBER de Enfermedades Raras (CIBERER), Madrid, Spain. E-mails: [email protected]; [email protected]; [email protected]; [email protected]; [email protected]; [email protected] † Current address: Departamento de Bioquímica y Biología Molecular I, Facultad de Biología, Universidad Complutense de Madrid, Madrid, Spain *Corresponding author 2 ABSTRACT Endoglin (CD105) is an auxiliary membrane receptor of transforming growth factor- (TGF-) that interacts with type I and type II TGF- receptors and modulates TGF- signalling. Mutations in endoglin are involved in Hereditary Hemorrhagic Telangiectasia type I, a disorder characterized by cutaneous telangiectasias, epistaxis (nosebleeds) and major arteriovenous shunts, mainly in liver and lung. Endoglin is overexpressed in the tumor-associated vascular endothelium where it modulates angiogenesis. This feature makes endoglin a promising target for antiangiogenic cancer therapy. Recent studies on human and experimental models of carcinogenesis point to an important tumor cell-autonomous role of endoglin by regulating proliferation, migration, invasion and metastasis. These studies suggest that endoglin behaves as a suppressor of malignancy in experimental and human carcinogenesis. In this review, we evaluate the implication of endoglin in tumor development underlying studies developed in our laboratories in recent years. -

Transforming Growth Factor-Β: a Multifunctional Regulator of Cancer Immunity
cancers Review Transforming Growth Factor-β: A Multifunctional Regulator of Cancer Immunity 1, 1, 1 Vivian Weiwen Xue y , Jeff Yat-Fai Chung y, Cristina Alexandra García Córdoba , Alvin Ho-Kwan Cheung 1, Wei Kang 1 , Eric W.-F. Lam 2 , Kam-Tong Leung 3, Ka-Fai To 1, Hui-Yao Lan 4 and Patrick Ming-Kuen Tang 1,* 1 Department of Anatomical and Cellular Pathology, State Key Laboratory of Translational Oncology, The Chinese University of Hong Kong, Hong Kong 999077, China; [email protected] (V.W.X.); jeff[email protected] (J.Y.-F.C.); [email protected] (C.A.G.C.); [email protected] (A.H.-K.C.); [email protected] (W.K.); [email protected] (K.-F.T.) 2 Department of Surgery and Cancer, Imperial College London, Hammersmith Hospital Campus, London W12 0NN, UK; [email protected] 3 Department of Paediatrics, The Chinese University of Hong Kong, Shatin, Hong Kong 999077, China; [email protected] 4 Department of Medicine and Therapeutics, Li Ka Shing Institute of Health Sciences, The Chinese University of Hong Kong, Hong Kong 999077, China; [email protected] * Correspondence: [email protected] These authors contributed equally to this work. y Received: 24 September 2020; Accepted: 12 October 2020; Published: 23 October 2020 Simple Summary: Transforming growth factor beta (TGF-β) is a multifunctional cytokine that can restrict cancer onset but also promote cancer progression at late stages of cancer. The ability of TGF-β in producing diverse and sometimes opposing effects relies on its potential to control different cellular signalling and gene expression in distinct cell types, and environmental settings. -

Lack of Tgfbr1 and Acvr1b Synergistically Stimulates Myofibre Hypertrophy And
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.03.433740; this version posted March 6, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Lack of Tgfbr1 and Acvr1b synergistically stimulates myofibre hypertrophy and accelerates muscle regeneration *M.M.G. Hillege1, *A. Shi1,2 ,3, R.C. Galli Caro1, G. Wu4, P. Bertolino5, W.M.H. Hoogaars1,6, R.T. Jaspers1 1. Laboratory for Myology, Department of Human Movement Sciences, Faculty of Behavioural and Movement Sciences, Vrije Universiteit Amsterdam, Amsterdam Movement Sciences, Amsterdam, The Netherlands 2. Department of Oral and Maxillofacial Surgery/Pathology, Amsterdam UMC and Academic Center for Dentistry Amsterdam (ACTA), Vrije Universiteit Amsterdam (VU), Amsterdam Movement Sciences (AMS), Amsterdam, the Netherlands 3. Key Laboratory of Oral Medicine, Guangzhou Institute of Oral Disease, Affiliated Stomatology Hospital of Guangzhou Medical University, Guangzhou Medical University, Guangzhou, China 4. Department of Oral Implantology and Prosthetic Dentistry, Academic Centre for Dentistry Amsterdam (ACTA), University of Amsterdam (UvA) and Vrije Universiteit Amsterdam (VU), The Netherlands 5. Centre de Recherche en Cancérologie de Lyon, UMR INSERM U1052/CNRS 5286, Université de Lyon, Centre Léon Bérard, Lyon, France 6. European Research Institute for the Biology of Ageing (ERIBA), University Medical Center Groningen (UMCG), University of Groningen, Groningen, The Netherlands *Contributed equally to this manuscript **Correspondence: [email protected]; Tel.: +31 (0) 205988463 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.03.433740; this version posted March 6, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

The Role of TGF-Β Superfamily Signaling in Neurological Disorders Risa Kashima1 and Akiko Hata1,2,*
Acta Biochim Biophys Sin, 2017, 1–15 doi: 10.1093/abbs/gmx124 Review Review The role of TGF-β superfamily signaling in neurological disorders Risa Kashima1 and Akiko Hata1,2,* 1Cardiovascular Research Institute, University of California, San Francisco, CA 94143, USA, and 2Department of Biochemistry and Biophysics, University of California, San Francisco, CA 94143, USA *Correspondence address. Tel: +1-415-476-9758; Fax: +1-415-514-1173; E-mail: [email protected] Received 12 September 2017; Editorial Decision 1 November 2017 Abstract The TGF-β superfamily signaling is involved in a variety of biological processes during embryo- genesis and in adult tissue homeostasis. Faulty regulation of the signaling pathway that trans- duces the TGF-β superfamily signals accordingly leads to a number of ailments, such as cancer and cardiovascular, metabolic, urinary, intestinal, skeletal, and immune diseases. In recent years, a number of studies have elucidated the essential roles of TGF-βs and BMPs during neuronal development in the maintenance of appropriate innervation and neuronal activity. The new advancement implicates significant roles of the aberrant TGF-β superfamily signaling in the patho- genesis of neurological disorders. In this review, we compile a number of reports implicating the deregulation of TGF-β/BMP signaling pathways in the pathogenesis of cognitive and neurodegen- erative disorders in animal models and patients. We apologize in advance that the review falls short of providing details of the role of TGF-β/BMP signaling or mechanisms underlying the patho- genesis of neurological disorders. The goal of this article is to reveal a gap in our knowledge regarding the association between TGF-β/BMP signaling pathways and neuronal tissue homeosta- sis and development and facilitate the research with a potential to develop new therapies for neurological ailments by modulating the pathways. -
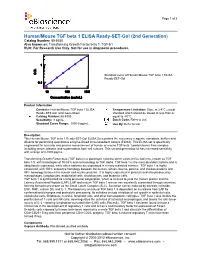
Human/Mouse TGF Beta 1 ELISA Ready-SET-Go! (2Nd Generation)
Page 1 of 3 Human/Mouse TGF beta 1 ELISA Ready-SET-Go! (2nd Generation) Catalog Number: 88-8350 Also known as: Transforming Growth Factor beta 1, TGF-b1 RUO: For Research Use Only. Not for use in diagnostic procedures. Standard curve of Human/Mouse TGF beta 1 ELISA Ready-SET-Go! Product Information Contents: Human/Mouse TGF beta 1 ELISA Temperature Limitation: Store at 2-8°C except Ready-SET-Go! (2nd Generation) standard which should be stored at less than or Catalog Number: 88-8350 equal to -70°C. Sensitivity: 8 pg/mL Batch Code: Refer to vial Standard Curve Range: 1000-8 pg/mL Use By: Refer to vial Description This Human/Mouse TGF beta 1 Ready-SET-Go! ELISA Set contains the necessary reagents, standards, buffers and diluents for performing quantitative enzyme-linked immunosorbent assays (ELISA). This ELISA set is specifically engineered for accurate and precise measurement of human or mouse TGF beta 1 protein levels from samples including serum, plasma, and supernatants from cell cultures. This second generation kit has increased sensitivity with a range of 8-1000 pg/mL. Transforming Growth Factor beta (TGF beta) is a pleiotropic cytokine which exists in five isoforms, known as TGF beta 1-5, with homologies of 70-80% and no homology to TGF alpha. TGF beta 1 is the most abundant isoform and is ubiquitously expressed, while other isoforms are expressed in a more restricted manner. TGF beta 1 is highly conserved, with 100% sequence homology between the human, simian, bovine, porcine, and chicken proteins and 99% homology between the human and murine proteins. -

Activemax® Recombinant Human TGF-Beta 1 / TGFB1 Catalog # AMS.TG1-H4212 for Research and Further Cell Culture Manufacturing Use
ActiveMax® Recombinant Human TGF-Beta 1 / TGFB1 Catalog # AMS.TG1-H4212 For Research and Further Cell Culture Manufacturing Use Description Source ActiveMax® Recombinant Human TGF-Beta 1 / TGFB1 (ActiveMax® Human TGF-Beta 1) Ala 279 - Ser 390 (Accession # NP_000651.3) was produced in human 293 cells (HEK293) Predicted N-terminus Ala 279 Molecular Characterization Endotoxin Less than 1.0 EU per μg of the ActiveMax® Human TGF-Beta 1 by the LAL method. Purity >95% as determined by SDS-PAGE of reduced (+) and non-reduced (-) rhTGFB1. Bioactivity The bio-activity was determined by its ability to inhibit IL-4 induced HT-2 cell proliferation. The ED50<0.05 ng/mL, corresponding to a specific activity of >2X107 Unit/mg Formulation and Storage Formulation Lyophilized from 0.22 μm filtered solution in TFA and acetonitrile. Normally Mannitol or Trehalose are added as protectants before lyophilization. Contact us for customized product form or formulation. Reconstitution See Certificate of Analysis for reconstitution instructions and specific concentrations. Storage Lyophilized Protein should be stored at -20℃ or lower for long term storage. Upon reconstitution, working aliquots should be stored at -20℃ or -70℃. Avoid repeated freeze-thaw cycles. No activity loss was observed after storage at: ● 4-8℃ for 12 months in lyophilized state; ● -70℃ for 3 months under sterile conditions after reconstitution. Background Background Transforming growth factor beta 1 ( TGFB1) is also known as TGF-β1, CED, DPD1, TGFB. is a polypeptide member of the transforming growth factor beta superfamily of cytokines. It is a secreted protein that performs many cellular functions, including the control of cell growth, cell proliferation, cell differentiation and apoptosis. -

TGF-B1 Suppresses Apoptosis Via Differential Regulation of MAP Kinases and Ceramide Production
Cell Death and Differentiation (2003) 10, 516–527 & 2003 Nature Publishing Group All rights reserved 1350-9047/03 $25.00 www.nature.com/cdd TGF-b1 suppresses apoptosis via differential regulation of MAP kinases and ceramide production H-H Chen1, S Zhao1 and J-G Song*,1 Introduction 1 Laboratory of Molecular Cell Biology, Institute of Biochemistry and Cell Biology, The balance between cell proliferation, differentiation, and Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, apoptosis is controlled by various internal and external stimuli, Shanghai, People’s Republic of China which play crucial roles in normal development and home- * Corresponding author: J Song, Laboratory of Molecular Cell Biology, Institute ostasis of living cells and organisms. Disruption of this of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, balance by different factors may lead to abnormal cell death, Chinese Academy of Sciences, 320 Yue-Yang Road, Shanghai 200031, growth, and many pathological events, including cancer, People’s Republic of China. E-mail: [email protected] immune and developmental diseases. Studies on the under- lying mechanisms through which the balance between cell Received 22.7.02; revised 18.10.02; accepted 29.10.02 Edited by JYJ Wang proliferation, differentiation, and apoptosis are maintained or disrupted are important in understanding various physiologi- cal and pathological processes. It is now known that complex Abstract signaling network, which is formed by the interaction or ‘crosstalk’ among various signaling molecules and pathways, Serum deprivation induces apoptosis in NIH3T3 cells, which is implicated in the subtle and general control of cell is associated with increased intracellular ceramide genera- proliferation, differentiation, and apoptosis. -

The Potential Role of Transforming Growth Factor Beta Family Ligand Interactions in Prostate Cancer
AIMS Molecular Science, 4(1): 41-61. DOI: 10.3934/molsci.2017.1.41 Received 19 October 2016, Accepted 25 January 2017, Published 13 February 2017 http://www.aimspress.com/journal/Molecular Review The potential role of transforming growth factor beta family ligand interactions in prostate cancer Kit P. Croxford, Karen L. Reader, and Helen D. Nicholson* Department of Anatomy, University of Otago, Dunedin 9054, New Zealand * Correspondence: Email: [email protected]; Tel: +64 3 479 5295. Abstract: The transforming growth factor beta (TGF-β) family plays an important role in embryonic development and control of the cell cycle. Members of the TGF-β family have pleiotropic functions and are involved in both the inhibition and progression of various cancers. In particular, deregulation of the TGF-β family has been associated with prostate cancer, as both a mechanism of disease progression and a possible therapeutic target. This review concentrates on the TGF-βs, activins and inhibins, bone morphogenetic proteins and NODAL and their connection to prostate cancer. Whilst most studies examine the family members in isolation, there are multiple interactions that may occur between members which can alter their function. Such interactions include ligand competition for receptor binding and shared intracellular pathways such as the Mothers against decapentaplegic (SMAD) proteins. Another mechanism for interaction within the TGF-β family is facilitated by their dimeric structure; heterodimers can form which exhibit different functional capabilities to their homodimeric counterparts. The potential formation of TGF-β family heterodimers has not been well examined in prostate cancer. The multiple methods of interrelations between members highlights the need for gross analysis of the TGF-β family and related factors in association with prostate cancer, in order to discover possible future avenues for TGF-β based diagnosis and treatments of the disease. -

Tgfβ/BMP Signaling Pathway in Cartilage Homeostasis
cells Review TGFβ/BMP Signaling Pathway in Cartilage Homeostasis Nathalie G.M. Thielen , Peter M. van der Kraan and Arjan P.M. van Caam * Experimental Rheumatology, Radboud University Medical Center, Geert Grooteplein 28, 6525 GA Nijmegen, The Netherlands * Correspondence: [email protected]; Tel.: +31-24-10513; Fax: +31-24-3540403 Received: 2 July 2019; Accepted: 19 August 2019; Published: 24 August 2019 Abstract: Cartilage homeostasis is governed by articular chondrocytes via their ability to modulate extracellular matrix production and degradation. In turn, chondrocyte activity is regulated by growth factors such as those of the transforming growth factor β (TGFβ) family. Members of this family include the TGFβs, bone morphogenetic proteins (BMPs), and growth and differentiation factors (GDFs). Signaling by this protein family uniquely activates SMAD-dependent signaling and transcription but also activates SMAD-independent signaling via MAPKs such as ERK and TAK1. This review will address the pivotal role of the TGFβ family in cartilage biology by listing several TGFβ family members and describing their signaling and importance for cartilage maintenance. In addition, it is discussed how (pathological) processes such as aging, mechanical stress, and inflammation contribute to altered TGFβ family signaling, leading to disturbed cartilage metabolism and disease. Keywords: transforming growth factor β; bone morphogenetic proteins; osteoarthritis; cartilage; SMADs; aging; joint loading; inflammation; linker modifications 1. Introduction The transforming growth factor β (TGFβ) family of polypeptide growth factors controls development and homeostasis of many tissues, including articular cartilage. Articular cartilage is the connective tissue covering joint surfaces and is a type of hyaline cartilage. This tissue is key in facilitating movement with its smooth lubricated surface, and it functions as a shock absorber to disperse forces acting upon movement with its physical properties.